Our previous studies documented that chronic NAC treatment certainly reduced the levels of reactive oxygen species in several forms of experimental hypertension including SHR (6-8). In NAC-treated animals we have observed decreased aortic lucigenin chemiluminescence, the changes of which correlated with the reduction of conjugated diene levels in heart and kidney (8). Reduction of conjugated diene levels was also associated with attenuated expression of redox-sensitive factor NF-
There is a considerable evidence that sympathetic hyperactivity in genetic hypertension results from altered balance between angiotensin II and nitric oxide in the brain, influencing the neural output from vasomotor centers in the brainstem (10-12). It seems that central nervous NO production in genetic hypertension is not capable to lower sufficiently the enhanced sympathetic tone driven by high activity of brain renin-angiotensin system (RAS). There are numerous contradictory findings concerning NO formation in the brainstem of SHR (13-17). Some of them indicate that enhanced expression and/or activity of neuronal NO synthase (nNOS) can at least partially counterregulate the increased sympathetic tone resulting from elevated brain angiotensin II levels (16, 18, 19). To our knowledge, the role of nNOS has never been analyzed using S-methyl-L-thiocitrulline (SMTC) which was reported to be a specific inhibitor of neuronal NOS (10-fold more potent against nNOS than eNOS and 17-fold more selective for rat nNOS in neuronal tissue than for rat eNOS in vascular endothelium) (20). Since SMTC had also peripheral effects, when administered in vivo (21), this nNOS inhibitor was chosen for the estimation of nNOS contribution in our in vitro assay of NOS activity in brain homogenates.
The aims of the present study were 1) to evaluate the effect of chronic NAC treatment on NOS activity during the development of high blood pressure in young SHR, and 2) to estimate the contribution of neuronal NO synthase (nNOS) to possible strain differences as well as to NAC-induced changes in the total NOS activity in brainstem and cerebellum of these animals. To achieve the second goal of our study we determined NOS activity also in the presence of a specific nNOS inhibitor S-methyl-thiocitrulline (SMTC).
Animals and treatment
All procedures and experimental protocols were approved by the Ethical Committee of the Institute of Physiology AS CR, and conform to the European Convention on Animal Protection and Guidelines on Research Animal Use. All the chemicals used were purchased from Sigma Chemicals Co. (Germany) except of [3H]-L-arginine (Amersham, UK).
Young 4-week-old male WKY (n=12) and SHR (n=15) were randomly divided into water drinking control groups and groups receiving N-acetylcysteine (NAC, 1.5 g/kg/day, 20 g/l) in tap water for 8 weeks. All animals were housed in the room with a stable temperature of 23±1°C and fed a regular pellet diet ad libitum. At the end of treatment, the blood pressure was determined by direct puncture of carotid artery under a light ether anesthesia. Thereafter the animals were sacrificed and body weight (BW) and heart weight (HW) were determined. Brainstem and cerebellum were dissected (17) and used for determination of of total NOS activity.
Total NO synthase activity
Total NO synthase activity was determined in crude homogenates (Potter, teflon homogenizer) of the cerebellum and brainstem by measuring the formation of [3H]-L-citrulline from [3H]-L-arginine as previously described by Bredt and Snyder (22) with minor modifications (23). Briefly, 50 µl of crude homogenate of the brain part (7.5 mg of wet tissue) was incubated in the presence of 50 mmol/l Tris/HCl, pH 7.4, containing 1 µmol/l [3H]-L-arginine (specific activity 5 GBq/mmol, approx. 100000 d.p.m.), 0.5 mg/ml calmodulin, 0.5 mmol/l ß-NADPH, 250 µmol/l tetrahydrobiopterin, 4 µmol/l FAD, 4 µmol/l flavin mononucleotide and 1 mmol/l Ca2+, in a total volume of 100 µl. Aliquots of the homogenate were also incubated in the presence of SMTC (10-6 mol/l). Fig. 1 shows dose-dependent inhibition of cerebellar NOS activity by increasing SMTC concentrations obtained in our preliminary experiments carried out in Wistar rats. The above concentration of SMTC has been chosen to demonstrate its predominant effect on nNOS because 10-5 mol/l SMTC seem to inhibit also the activity of other NOS isoforms (20). After a 30 min incubation at 37°C, the reaction was stopped (by adding 0.02 M Hepes containing 2 mM EDTA, 2 mM EGTA and 1 mM L-citrulline), the samples were centrifuged, and supernatants were applied to 1 ml Dowex 50WX-8 columns (Na+ form). L-citrulline was eluted with 2 ml of water and [3H]-L-citrulline radioactivity was determined by liquid scintillation counting. Total NO synthase activity was expressed as pkat/g of proteins.
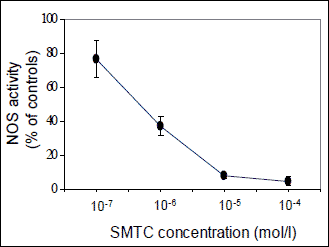 |
Fig. 1. Dose-dependent inhibition of NO synthase (NOS) activity by S-methyl-L-thiocitrulline (SMTC) in the homogenate of cerebellum from Wistar rats. Data are means±S.E.M. (n=4). |
Statistical analysis
Results were expressed as means ± SEM. The data were evaluated by means of one-way analysis of variance with Fisher least significant difference (LSD) post-hoc test. P<0.05 value was taken as a criterion of significant relationship.
SHR were characterized by increased blood pressure and higher relative heart weight compared to WKY controls (Table 1). Total NOS activity was increased in brainstem of SHR compared to WKY (4.4±0.4 vs. 3.3±0.1 pkat/g proteins, p<0.05), but this difference was not significant in the cerebellum (5.8±0.6 vs. 5.0±0.7 pkat/g proteins). These strain differences were due to the changes in SMTC-sensitive NOS activity, whereas no strain differences in SMTC-insensitive NOS activity were observed (Figs. 2 and 3).
| Table 1. Blood pressure, body and heart weight in untreated and NAC-treated WKY and SHR rats |
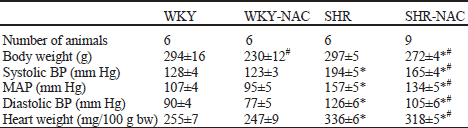 |
| Data are means ± S.E:M. MAP - mean arterial pressure. Significantly different (p<0.05): * from WKY, # effect of NAC treatment. |
Chronic NAC treatment reduced blood pressure and heart weight in SHR only (Table 1). Chronic NAC treatment increased total NOS activity by 45% in the brainstem and by 38% in the cerebellum of Wistar-Kyoto rats. Similarly, NAC treatment of SHR increased total NOS activity by 32% in the brainstem and by 67% in the cerebellum. This effect was due to the rise of SMTC-sensitive activity. In contrast, no significant change in SMTC-insensitive NOS activity was found in brainstem or cerebellum of both strains (Figs. 2 and 3). Thus, in SHR SMTC inhibited 86% and 70% of NAC-induced increase of total NOS activity in the brainstem and cerebellum, respectively. Taken together, nNOS is responsible for increased total NOS activity in the brain of SHR as well as for NAC-induced changes of NOS activity.
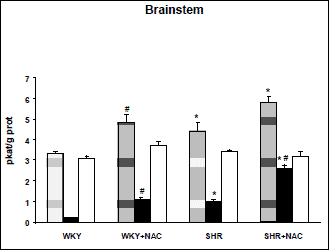 |
Fig. 2. Nitric oxide synthase (NOS) activity in the brainstem of untreated and NAC-treated Wistar-Kyoto (WKY) and spontaneously hypertensive rats (SHR). Total NOS activity (hatched column), SMTC-sensitive NOS activity (black column), and SMTC-insensitive NOS activity (white column). Data are means±S.E.M. SMTC was used in 10-6 M concentration. Significantly different (p<0.05): *- from WKY, #- effect of NAC treatment. |
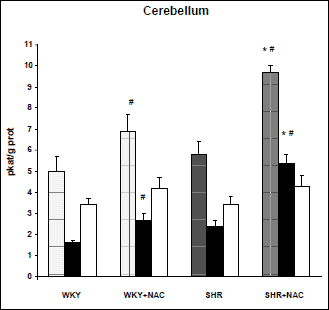 |
Fig. 3. Nitric oxide synthase (NOS) activity in the cerebellum of untreated and NAC-treated Wistar-Kyoto (WKY) and spontaneously hypertensive rats (SHR). Total NOS activity (hatched column), SMTC-sensitive NOS activity (black column), and SMTC-insensitive NOS activity (white column). Data are means±S.E.M. SMTC was used in 10-6 M concentration. Significantly different (p<0.05): *- from WKY, #- effect of NAC treatment. |
Our study demonstrated higher SMTC-sensitive NOS activity in the brainstem of 12-week-old SHR compared to age-matched WKY rats. This difference in neuronal NOS activity was further augmented by chronic N-acetylcysteine treatment, which lowered blood pressure in SHR but not in WKY rats. Chronic NAC treatment augmented SMTC-sensitive NOS activity not only in the brainstem but also in the cerebellum. This effect was also present in WKY brain but to a smaller extent compared to the changes seen in SHR brain. Neuronal NOS plays not only an important role in the central control of sympathetic tone (see below) but also in the modulation of the response of hypothalamo-pituitary-adrenal axis to a-adrenergic or b-adrenergic stimulation (24).
The mechanisms of N-acetylcysteine-induced increase of NO bioavaibility in the cardiovascular tissues and in the brain are not completely elucidated yet. NAC has been reported to posses potent antioxidant effects by both direct neutralization of reactive oxygen species (25) and increased GSH production (26). Using TEAC assay, the antioxidant activity of NAC was found to be comparable with the activity of Trolox (7). Furthermore, NAC treatment reduced NF-
These beneficial effects of NAC were described rather in the peripheral tissues. Nevertheless, antioxidant effects of NAC accompanied by improvement of NO pathway may operate within the brain as well. Recently, Arakawa and Ito (30) and Jayalakshmi et al. (31) described neuroprotective effect of NAC which includes redox interactions, protection against oxidative stress-induced damage and against hypoxia. Moreover, using the cerebrovascular arteries Sury et al. (32) demonstrated that NAC was able to inhibit endothelin-1 upregulation induced by TNF-alpha. Mitogen and stress-activated protein kinase was involved in this inhibitory effect. Improvement of NO pathway within the central nervous system may further modulate sympathetic stimulation of peripheral arteries. Macarthur et al. (33) documented that chronic NAC administration reduced nerve-stimulated norepinephrine overflow but increased neuropeptide Y overflow in the mesenteric arterial bed from the SHR. This property of NAC was prevented by inhibiting NO synthase with L-NAME demonstrating that the effect of NAC was due to the preservation of NO pathway.
Our present findings on NAC-induced augmentation of SMTC-sensitive NOS activity in the brain of SHR seem to be compatible with the suggested inhibitory role of brain NO in the central control of sympathetic tone (10, 34). It was demonstrated at the level of rostral ventrolateral medulla that NO counterbalances angiotensin II-mediated rise of sympathetic outflow (35-37). Chronic NAC treatment of young SHR attenuated the development of hypertension (6) and this effect was ascribed to antioxidant and NOS-stimulating effects of this treatment. Girouard et al. (38) reported that NAC improves nitric oxide vasodilatation and attenuates alpha-adrenergic vasoconstriction in mesenteric beds of spontaneously hypertensive rats. Our study in salt hypertensive Dahl rats (39) indicated that blood pressure lowering effect of chronic NAC treatment was almost exclusively due to a profound reduction of sympathetic vasoconstriction which is greatly enhanced in this hypertensive model. It seems that the antioxidant effects of chronic NAC treatment might be even more important for blood pressure reduction than NAC-induced stimulation of NOS activity because chronic administration of NO donor - pentaerythrityl tetranitrate - did not attenuate hypertension development in young SHR (40).
On the basis of the above findings, we can suggest that NAC-induced elevation of neuronal NOS activity in the brainstem of SHR might significantly contribute to the observed blood pressure decrease by the reduction of sympathetic tone in SHR. This is further combined with the improvement of endothelial dysfunction and NO-dependent vasodilatation in peripheral tissues which were also reported to be impaired in SHR (41).
Acknowledgements: This work was supported by AV0Z 5110509, by Centre for Cardiovascular Research (1M0510), by research grants of GA CR 305/08/0139, VEGA 2/0178/09 and APVV-0538-07.
Conflict of interests: None declared.
- Head R.J. Hypernoradrenergic innervation: its relationship to functional and hyperplastic changes in the vasculature of the spontaneously hypertensive rat. Blood Vessels 1989; 26: 1-20.
- De Champlain J. Pre- and postsynaptic adrenergic dysfunctions in hypertension. J Hypertens 1990 ; 8(Suppl 7): S77-S85.
- Luscher TF. Imbalance of endothelium-derived relaxing and contracting factors. A new concept in hypertension? Am J Hypertens 1990; 3: 317-330.
- Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 2004; 44: 245-252.
- Kojsova S, Jendekova L, Zicha J, Kunes J, Andriantsitohaina R, Pechanova O. The effect of different antioxidants on nitric oxide production in hypertensive rats. Physiol Res 2006; 55 (Suppl 1): S3-S16.
- Pechanova O, Zicha J, Kojsova S, Dobesova Z, Jendekova L, Kunes J. Effect of chronic N-acetylcysteine treatment on the development of spontaneous hypertension. Clin Sci 2006; 110: 235-242.
- Pechanova O, Zicha J, Paulis L, et al. The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur J Pharmacol 2007; 561: 129-136.
- Rauchova H, Pechanova O, Kunes J, Vokurkova M, Dobesova Z, Zicha J. Chronic
N-acetylcysteine administration prevents development of hypertension in
N
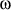 -nitro-L-arginine
methyl ester-treated rats: the role of reactive oxygen species. Hypertens
Res 2005; 28: 472-485.
-nitro-L-arginine
methyl ester-treated rats: the role of reactive oxygen species. Hypertens
Res 2005; 28: 472-485. - Zicha J, Dobesova Z, Kunes J. Antihypertensive mechanisms of chronic captopril or N-acetylcysteine treatment in L-NAME hypertensive rats. Hypertens Res 2006; 29: 1021-1027.
- Krukoff TL. Central regulation of autonomic function: NO brakes? Clin Exp Pharmacol Physiol 1998; 25: 474-478.
- Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: recent advances and perspectives. Br J Pharmacol 2003; 139: 191-202.
- Dampney RA, Horiuchi J, Killinger S, Sheriff MJ, Tan PS, McDowall LM. Long-term regulation of arterial blood pressure by hypothalamic nuclei: some critical questions. Clin Exp Pharmacol Physiol 2005; 32: 419-425.
- Cabrera CL, Bealer SL, Bohr DF. Central depressor action of nitric oxide is deficient in genetic hypertension. Am J Hypertens 1996; 9: 237-241.
- Kagiyama S, Tsuchihashi I, Abe I, Fujishima M. Enhanced depressor response to nitric oxide in the rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertension 1998; 31: 1030-1034.
- Qadri F, Arens T, Schwarz E-C, Hauser W, Dendorfer A, Dominiak P. Brain nitric oxide synthase activity in spontaneously hypertensive rats during the development of hypertension. J Hypertens 2003; 21: 1687-694.
- Hauser W, Sassmann A, Qadri F, Johren O, Dominiak P. Expression of nitric oxide synthase isoforms in hypothalamo-pituitary-adrenal axis during the development of spontaneous hypertension in rats. Brain Res Mol Brain Res 2005; 138: 198-204.
- Hojna S, Kadlecova M, Dobesova Z, Valouskova V, Zicha J, Kunes J. The participation of brain NO synthase in blood pressure control of adult spontaneously hypertensive rats. Mol Cell Biochem 2007; 297: 21-29.
- Plochocka-Zulinska D, Krukoff TL. Increased gene expression of neuronal nitric oxide synthase in brain of adult spontaneously hypertensive rats. Brain Res Mol Brain Res 1997; 48: 291-297.
- Edwards MA, Loxley RA, Powers-Martin K, et al. Unique levels of expression of N-methyl-D-aspartate receptor subunits and neuronal nitric oxide synthase in the rostral ventrolateral medulla of the spontaneously hypertensive rat. Brain Res Mol Brain Res 2004; 129: 33-43.
- Furfine ES, Harmon MF, Paith JE, et al. Potent and selective inhibition of human nitric oxide synthases. Selective inhibition of neuronal nitric oxide synthase by S-methyl-L-thiocitrulline and S-ethyl-L-thiocitrulline. J Biol Chem 1994; 269: 26677-26683.
- Wakefield ID, March JE, Kemp PA, Valentin JP, Bennett T, Gardiner SM. Comparative regional haemodynamic effects of the nitric oxide synthase inhibitors, S-methyl-L-thiocitrulline and L-NAME, in conscious rats. Br J Pharmacol 2003; 139: 1235-1243.
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 1990; 87: 682-685.
- Pechanova O, Bernatova I. Effect of captopril on cyclic nucleotide concentration during long-term NO synthase inhibition. Physiol Res 2000; 49: 55-63.
- Gadek-Michalska A, Bugajski J. Nitric oxide in the adrenergic- and CRH-induced activation of hypothalamo-pituitary-adrenal axis. J Physiol Pharmacol 2008; 59: 365-378.
- Martina V, Masha A, Gigliardi VR, et al. Long-term N-acetylcysteine and L-arginine administration reduces endothelial activation and systolic blood pressure in hypertensive patients with type 2 diabetes. Diabetes Care 2008; 31: 940-944.
- Cabassi A, Dumont EC, Girouard H, et al. Effect of chronic N-acetylcysteine treatment on the actions of peroxynitrate on aortic vascular reactivity in hypertensive rats. J Hypertens 2001; 19: 1233-1244.
- Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart : role of oxidative stress. Am J Pathol 2002; 161: 1773-1781.
- Lahera V, Khraibi AA, Romero JC. Sulfhydryl group donors potentiate the hypotensive effect of acetylcholine in rats. Hypertension 1993; 30: 934-941.
- Brozmanova M, Plevka J, Bartos V, et al. Oral N-acetylcysteine reverses hyperoxia-induced cough suppression in guinea pigs. J Physiol Pharmacol 2008; 58(Suppl 5): 75-84.
- Arakawa M, Ito Y. N-acetylcysteine and neurodegenerative diseases: basic and clinical pharmacology. Cerebellum 2007; 19: 1-7.
- Jayalakshmi K, Sairam M, Singh SB, Sharma SK, Ilavazhagan G, Banerjee PK. Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress in primary hippocampal culture. Brain Res 2005; 1046: 97-104.
- Sury MD, Frese-Schaper M, Muhlemann MK, et al. Evidence that N-acetylcysteine inhibits TNF-alpha-induced cerebrovascular endothelin-1 upregulation via inhibition of mitogen- and stress-activated protein kinase. Free Radic Biol Med 2006; 41: 1372-1383.
- Macarthur H, Westfall TC, Wilken GH. Oxidative stress attenuates NO-induced modulation of sympathetic neurotransmission in the mesenteric arterial bed of spontaneously hypertensive rats. Am J Physiol 2008; 294: H183-H189.
- Osborn JW. Hypothesis: set-points and long-term control of arterial pressure. A theoretical argument for a long-term arterial pressure control system in the brain rather than the kidney. Clin Exp Pharmacol Physiol 2005; 32: 384-393.
- Bergamaschi CT, Campos RR, Lopes OU. Rostral ventrolateral medulla: a source of sympathetic activation in rats subjected to long-term treatment with L-NAME. Hypertension 1999; 34: 744-747.
- Tsuchihashi T, Kagiyama S, Matsumura K, Lin Y, Abe I, Fujishima M. Cardiovascular responses to glutamate and angiotensin II in ventrolateral medulla of hypertension induced by chronic inhibition of nitric oxide. Hypertens Res 2000; 23: 359-364.
- Bergamaschi CT, Biancardi VC, Lopes OU, Campos RR. Effects of angiotensin blockade in the rostral ventrolateral medulla on maintenance of hypertension induced by chronic L-NAME treatment. Brain Res 2002; 927: 195-199.
- Girouard H, Chulak C, Wu L, LeJossec M, de Champlain J. N-Acetylcysteine improves nitric oxide and alpha-adrenergic pathways in mesenteric beds of spontaneously hypertensive rats. Am J Hypertens 2003; 16: 577-584.
- Kunes J, Dobesova Z, Zicha J. Chronic N-acetylcysteine treatment prevents the development of salt hypertension in immature Dahl rat. J Hypertens 2004; 22(Suppl 2): S336.
- Kristek F, Koprdova R, Cebova M. Long-term effects of early administered sildenafil and NO donor on the cardiovascular system of SHR. J Physiol Pharmacol 2007; 58: 33-43.
- Bernatova I, Csizmadiova Z, Kopincova J, Puszerova A. Vascular function and nitric oxide production in chronic social-stress-exposed rats with various family history of hypertension. J Physiol Pharmacol 2007; 58: 487-501.