WEIGHT LOSS AND EXERCISE
It is well accepted that simple lifestyle modifications such as body weight management with appropriate nutritional counseling, with or without regular physical exercise and cognitive-behavior programs should be the mainstays of therapy for patients with metabolic syndrome. It has been shown that obesity together with insulin resistance, type 2 diabetes and dyslipidemia is a central risk factor for NAFLD, and visceral adipose tissue has an important role in secretion of several adipokines and cytokines which cause systemic and hepatic insulin resistance, as well as hepatocyte injury and apoptosis, neutrophil chemotaxis, and hepatic stellate cell activation (9, 10). Body weight reduction on the other hand leads to the loss of the adipose tissue, which further leads to the improvement in peripheral and hepatic insulin sensitivity and prevention of hepatic injury. Specific diet and exercise guidelines for NAFLD patients havent been published, but there are several types of diets which have been proposed for treatment of obesity and metabolic syndrome by different medical and commercial sources (11-19). Most of the published studies in NAFLD population have been consistent with notion that gradual weight loss with a calorie-restricted diet and concomitant exercise leads to a decrease in the incidence of metabolic syndrome, improvement in liver biochemical tests, and resolution of hepatic steatosis (20-24). Rapid weight loss can however aggravate the underlying liver disease causing the portal inflammation and fibrosis, and moderate and graduate weight loss not exceeding loss of 1.6 kg/week with incorporated regular physical aerobic activity regimen including at least 30 min of exercise three times per week is therefore advocated (25). According to the published results, the weight reduction of 5% or more accompanied by regular exercise for one year has been associated with improvement and normalization in liver tests, and keeping this weight (<5% gain) for two consecutive years maintained normal ALT levels (26). Huang et al. showed that intense dietary intervention can achieve a reduction in mean waist circumference, insulin resistance, levels of fasting glucose, triglycerides and liver function tests, as well as an improvement in liver histology in patients with NASH (27). Home-based lifestyle modification intervention carried out in collaboration with interdisciplinary medical staff should be the preferred approach in obese NAFLD patients (28). However, there is a problem that the positive effect is commonly short lived, implementation of these lifestyle changes is not always successful, particularly in very obese patients, and long-term data on the liver histological improvement are still lacking. In contrary to the most of published results, study by Larson-Meyer et al. showed that there is significant reduction of liver lipid content (measured by magnetic resonance spectroscopy) with caloric restriction (with or without exercise), but without significantly correlated reduction in triglycerides, high-density lipoprotein (HDL)-cholesterol, or ALT levels as well as markers of systemic inflammation (29). Pharmacological treatment for obesity may be offered to those patients with a BMI greater than 30 kg/m2 who had no change in the course of disease after adequate lifestyle changes have been undertaken. Orlistat (inhibitor of pancreatic and gastric lipase), was evaluated in a pilot study which included ten obese NASH patients treated for 6 months with this agent (30). Results showed that 10% or greater consequent body weight reduction lead to improvement in aminotransferase levels, liver steatosis and fibrosis. In another double-blind randomized placebo-controlled trial group of patients with NAFLD treated with orlistat for 6 months had significant 2-fold reduction of serum ALT levels and a statistically significant reversal of liver steatosis detected by ultrasound (31). Third study in obese NAFLD patients confirmed a histological improvement in liver fibrosis and inflammation after treatment with orlistat as well as improvement in insulin resistance index and reduction of aminotransferases, total cholesterol, triglycerides and low-density lipoprotein levels (32). Sibutramine is a specific inhibitor of norepinephrine, serotonin and dopamine reuptake into nerve terminals which leads to increased satiety, inhibition of food intake and possibly elevation in energy expenditure. A 6-month study of sibutramine in the obese subjects with NASH and concomitant low calorie diet showed reduction of body weight and insulin resistance, with improvements in biochemical markers and ultrasound findings of NASH (33). Results of these studies are interesting but there are few concerns which should be addressed. Long-term safety profile of both of these agents is unknown. Orlistat causes gastrointestinal side-effects and malabsorption of fat-soluble vitamins in up to 30% of patients, and sibutramine can increase blood pressure levels (34, 35). Although the mechanisms of action of these two agents do not overlap, their combination is not recommended because there is no proven additive weight loss when used together, and the rate of combined side-effects is unacceptable (36). Bariatric surgery offers advantage of effective long-term weight management in patients with severe obesity. It is associated with improvement in diabetes mellitus, hypertriglyceridemia, hypertension and obstructive sleep apnea and is possible the best therapeutic modality in NAFLD patients with severe obesity (37, 38). Older or classical procedures such as jejuno-ileal bypass have been almost completely replaced by newer procedures such as proximal gastric bypass and biliopancretic diversion. The reason is the high rate of postoperative complications reported with classical procedures, even though cases of NASH progression and subacute liver failure have also been reported with newer methods (39, 40). Improvement in diabetes mellitus, liver function tests and liver histology (inflammation and fibrosis) was reported in several trials of severe obese NASH patients treated with gastroplasty (41-44), and similar results were reported after gastric bypass surgery (45). Nevertheless, the decision when and in which patients to perform bariatric surgery should still be highly individual. Liposuction is nowadays a commonly used procedure for removal of subcutaneous fat tissue. It offers the advantage of significant weight and fat loss but does not improve peripheral insulin sensitivity and doesnt affect the levels of adipokines (46). Therefore it cannot be recommended as a treatment modality for obese NAFLD patients. Specific recommendations regarding smoking and minimal to moderate alcohol intake in patients with NAFLD cannot be made clearly. On the other hand, cessation of smoking should be one of the aims of primary prevention of metabolic syndrome conditions with an increased risk of mortality, especially in patients at a high risk for cardiovascular disease (47-48).
Metformin is a biguanide, a class of oral hypoglycemic drugs with insulin-sensitizing properties which acts through decreasing the hepatic glucose output, increasing the insulin-mediated glucose utilization in peripheral tissues, and lowering the serum free fatty acid concentrations (49-51). It is considered as the first choice in oral treatment of type 2 diabetes for several reasons: it promotes modest weight reduction in contrast to other hypoglycemic agents, it is less likely to cause hypoglycemia, it has a lipid-lowering activity, and has also a low market cost (52). Animal studies have reported that metformin reverses hepatomegaly, steatosis and liver tests abnormalities in animal models of NAFLD (53). Several smaller clinical studies initially reported improvement in mean serum aminotransferase levels and insulin resistance after 6 months of treatment; although after 1 year of treatment there was no clear effect on aminotransferase levels, liver histology, or insulin sensitivity (54-56). In an open-label randomized study of 110 non-diabetic NAFLD patients, higher rates of ALT normalization and improvement in liver histology and insulin sensitivity were associated with metformin treatment in comparison to vitamin E treatment or weight loss (57). However, some recent open-label randomized studies have found no benefit of metformin treatment on liver steatosis, assessed either histologically or by CT, aminotransferase levels, or markers of insulin resistance and inflammation in comparison to placebo or lifestyle interventions (58-61). Therefore, it cannot be recommended as an initial treatment for non-obese or obese NAFLD patients, although it still has a role in a treatment of group of NAFLD patients with impairment in glucose or lipid metabolism.
Thiazolidinediones (pioglitazone, rosiglitazone) are novel class of oral antidiabetic drugs that ameliorate insulin sensitivity in euglycemic and diabetic patients by activating peroxisome proliferator-activated receptor (PPAR) gamma (62). In a recent animal study, it has been demonstrated that preventive pioglitazone effects in NASH depend on adiponectin upregulation and subsequent inhibition of sterol regulatory element-binding protein 1c (SREBP-1c), which controls the expression of nearly all genes required for de novo synthesis of fatty acids and triglyceride synthesis (63). Adiponectin has been identified as a pivotal mediator for thiazolidinediones effects on glucose homeostasis, insulin sensitivity and lipid metabolism (64). It increases fatty acid ß-oxidation in muscle, decreases hepatic lipid content in ob/ob mice and has direct anti-fibrotic and anti-inflammatory properties (65-70). In studies with animals, pioglitazone and rosiglitazone prevented activation of hepatic stellate cells in vitro and improved hepatic steatosis and prevented liver fibrosis in vivo (71, 72). In another animal study, 1 week treatment with pioglitazone significantly decreased hepatic triglyceride content and serum levels of TNF-
Given the fact that hypertriglyceridemia and low HDL levels are defining elements of metabolic syndrome, and that those patients with elevated serum LDL-cholesterol and established coronary disease with other characteristics of the metabolic syndrome have the highest risk of major coronary events it was expected in course of time that studies exploring role of lipid-lowering agents in treatment of patients with NAFLD will be undertaken (84-86). A one year pilot study of clofibrate in the treatment of patients with hypertriglyceridemia and NASH, revealed no significant changes in levels of aminotransferases or liver histology (87). On the other hand results from a controlled trial of a four week treatment in patients with NASH, using gemfibrozil 600 mg/d showed significant biochemical improvements, but without histological confirmation (88). Three separate small pilot trails using HMG-CoA reductase inhibitor atorvastatin (10-80 mg/d) for 6-12 months have shown significant improvement or normalization in serum aminotransferase and lipids/cholesterol levels as well as improvement in hepatic inflammation in patients with NAFLD (89-91). Similar results were obtained by small pilot study using another HMG CoA reductase inhibitor pravastatin but without improvement in the fibrosis score (92). Greek open-label, randomised study in 186 non-diabetic patients with metabolic syndrome reported that combination treatment (atorvastatin+fenofibrate) combined with treatment of other components of metabolic syndrome (hypertension, impaired fasting glucose, and obesity) led to resolution of biochemical plus ultrasonographic evidence of NAFLD (93). A cumulative histopathological follow-up class study evaluated effect of statin therapy on histological outcome in patients with NAFLD (94). A significant reduction of liver steatosis at follow-up was noted in group of NAFLD patients taking statins. Recent animal study evaluated a novel combination therapy for NAFLD comprising ezetimibe, a cholesterol absorption inhibitor, and acarbose, an alpha-glucosidase inhibitor (95). Twenty four week treatment with combination therapy significantly reduced liver steatosis, inflammation and fibrosis, compared with long-term monotherapy with either drug. It also led to increase in the expression of microsomal triglyceride transfer protein and PPAR-
There is a certain role for angiotensin receptor blockers (ARB) in treatment of NAFLD patients. Renin-angiotensin system (RAS) has an important role in insulin resistance; suppressing the RAS improves intracellular insulin signaling through activation of PPAR-gamma, improves control of adipokines production, and prevents hepatic stellate cell activation which consequently leads to prevention of hepatic inflammation and fibrogenesis (102). Therefore studies with losartan were performed in patients with NASH and arterial hypertension (103-105). Biochemical improvement with reduction in levels of blood markers of fibrosis, TGF-ß1 and ferritin was reported. Also, after 48 weeks of treatment histological improvement was detected in majority of liver biopsy specimens. Because of these promising initial results further studies were performed in NAFLD patients. Enjoji et al. reported that telmisartan and olmesartan improve insulin sensitivity (HOMA-IR) and ALT levels and could be used as liver protecting agents in NAFLD patients (106). Romanian authors have compared efficacy of telmisartan vs. valsartan in 54 patients with NASH and mild-to-moderate hypertension, treated for 20 months (107). Telmisartan was superior according to the therapy induced mean HOMA-IR decrease per month, and reduction of NASH score. Recent animal study reported that 12 weeks of losartan treatment strongly reduces hepatic PAI-1 gene expression which could consequently lead to prevention of fatty liver disease (108). Another animal study gave us some more insights on the efficacy and mechanisms of action of telmisartan (109). It attenuates liver steatosis and fibrogenesis with decreased type I collagen and transforming growth factor-beta1 mRNA expressions and also suppresses the infiltration of macrophages into the liver, reduces the size of adipocyte in visceral fat tissue with an elevation of serum adiponectin concentration. Considering the standard and wide use of ARB in treatment of arterial hypertension, a component of metabolic syndrome, further studies in NAFLD patients should be encouraged.
Ursodeoxycholic acid (UDCA), a hydrophilic bile acid, acts by competitive displacement of hepatotoxic hydrophobic endogenous bile acids that facilitate apoptosis, minimizing their toxicity and leading to a decrease in oxidative stress and hepatic injury (110). Several initial smaller studies have suggested potential benefit of UDCA treatment in patients with NASH alone or in combination with low-fat diet or lipid lowering agents (87, 90, 111-113). Improvements in liver enzymes levels, degree of liver steatosis and serum markers of fibrosis suggested a possible beneficial effect of UDCA in NASH patients. However, subsequent large randomized double-blind placebo-controlled trial involving 166 biopsy-proven NASH patients who were randomized to UDCA 13-15 mg/kg/day or placebo for 2 years did not show any significant difference in liver enzymes improvement or histology between the two study groups (114). Dufour et al. reported improved biochemistry and histology, mostly due to regression of hepatic steatosis, in biopsy-proven NASH patients treated with UDCA and vitamine E compared to UDCA alone or placebo (115). Recent study suggests that UDCA+vitamin E combination treatment also has metabolic effects, in addition to its beneficial cytoprotective properties (116). Improvement in terms of liver biochemistry and histology (of patients with NASH) was also followed by a decreased hepatocellular apoptosis and increased circulating levels of adiponectin.
Vitamin E (a-tocopherol) is an antioxidant that stabilizes biological membranes by protecting unsaturated fatty acids from lipid peroxidation and subsequent free radical reaction (110). A relation between hepatic fibrosis and TGF-ß1 and pivotal role of TGF-ß1 in hepatic fibrogenesis and hepatocyte apoptosis has been reported (117-119). Vitamin E inhibits hepatic TGF-ß1 gene expression and subsequently attenuates cytokine stimulation of stellate cells and protects against liver fibrosis (120). Study by Hasegawa et al. with vitamin E administration (300 mg/day during 1 year) showed significant decrease in plasma TGF-ß1 levels along with normalization in liver enzymes and improvement in steatosis, inflammation and fibrosis on follow-up liver biopsy (121). Another small pilot study found that vitamin E administration normalizes liver tests (122). Subsequently, two small randomized controlled trials failed to provide any benefit of vitamin E (123, 124). A double blind, randomized, placebo-controlled trial that included 45 patients comparing vitamin E (1000 IU/day) and vitamin C (1000 mg/day) with placebo for 6 months showed no statistically significant difference in aminotransferase levels or degree of steatosis and inflammatory activity between vitamin E and placebo group, although an improvement in fibrosis score in vitamin E group was noted (125). In another study patients were randomized to vitamin E 600 IU/day plus ascorbic acid 500 mg/day (n =25) or placebo (n=28), and treated for 24 months (126). All patients also underwent lifestyle intervention (diet and increased physical activity). Although significant improvement occurred in the levels of aminotransferases along with grade of steatosis, lobular inflammation, hepatocyte ballooning, and NAFLD activity score, there was no significant difference in all these parameters between the two groups. Apparently, vitamin E plus ascorbic acid does not seem to increase the efficacy of lifestyle intervention alone. In recent animal study, vitamin E showed an antioxidant effect on the liver but it was not sufficient to improve liver histology (127). This suggests that vitamin E alone, characterized only with antioxidant properties, is insufficient to inhibit progression of NAFLD and combination with other therapeutic modalities are needed. A multicentered, randomized, placebo-controlled, double-blinded clinical trial comparing metformin with vitamin E or placebo in 173 nondiabetic children with histologically confirmed NAFLD will hopefully give more information about their effect on liver histology (128).
Polyunsaturated fatty acids (PUFA) are ligands of peroxisome proliferator-activated receptor
As it is postulated, adipokines and cytokines such as adiponectin, TNF-
According to the recent surveys, the traits of metabolic syndrome can be found in up to one-fourth to one-third of general population. It is therefore not surprising that NAFLD has become the most common chronic liver disease in Western countries, owing its rise primarily to the increasing prevalence of obesity in virtually all age groups. We are still pretty much unable to make a definitive diagnosis of NAFLD or to distinguish between NAFLD and NASH without liver biopsy, although recent advances through research of single nucleotide polymorphisms and development of noninvasive markers of hepatic fibrosis could improve the situation in the near future. NAFLD is a silent disease with slowly progressive nature which requires long-term follow-up. Most of the patients can be safely managed conservatively with a repeated check of liver tests without any need for further invasive work-up or medical treatment. Standard approach to these patients should be an effort to control and treat the potential risk factors such as obesity, hyperlipidemia, hypertension and diabetes. Weight reduction has clearly been associated with improvement in liver biochemical tests and prevention of further hepatic injury. It should be gradual, since rapid weight loss can aggravate underlying liver disease. Over the last two decades large numbers of pharmacologic agents have been studied as potential treatment options for NAFLD. However, the majority of studies have small sample sizes, short follow-up duration, and hence also limited statistical power, which makes it rather difficult to draw any definitive conclusions about the use of these agents. Only a few of described agents have been shown to have substantial effects and until today no single or combination therapy has come upfront in a way that it could be firmly recommended in the treatment of NAFLD. High prevalence and natural history potentially leading to end-stage liver disease clearly indicate the necessity for efficient therapy for a large number of these patients. We believe that firmer conclusions about NAFLD treatment will become available in the near future, mainly through diverse ongoing and forthcoming studies which address not only the potential treatment options, but also disease pathogenesis and possibility of categorization of patients with respect to disease prognosis.
Conflict of interests: None declared.
- Chitturi S, Farrell GC, Hashimoto E, Saibara T, Lau GK, Sollano JD. Non-alcoholic fatty liver disease in the Asia-Pacific region: definitions and overview of proposed guidelines. J Gastroenterol Hepatol 2007; 22: 778-787.
- Amarapurkar DN, Hashimoto E, Lesmana LA, Sollano JD, Chen PJ, Goh KL. How common is non-alcoholic fatty liver disease in the Asia-Pacific region and are there local differences? J Gastroenterol Hepatol 2007; 22: 788-793.
- Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003; 37: 917-923.
- Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413-1419.
- Day CP, James OF. Steatohepatitis: a tale of two hits. Gastroenterology 1998; 114: 842-845.
- Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis 2001; 21: 27-41.
- Tsochatzis EA, Papatheodoridis GV, Archimandritis AJ. Adipokines in nonalcoholic steatohepatitis: from pathogenesis to implications in diagnosis and therapy. Mediators Inflamm 2009; 2009: 831670.
- Ramadori G, Moriconi F, Malik I, Dudas J. Physiology and pathophysiology of liver inflammation, damage and repair. J Physiol Pharmacol 2008; 59(Suppl 1): 107-117.
- Duvnjak M, Lerotic I, Barsic N, Tomasic V, Virovic Jukic L, Velagic V. Pathogenesis and management issues for nonalcoholic fatty liver disease. World J Gastroenterol 2007; 13: 4539-4550.
- Younossi ZM. Review article: current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2008; 28: 2-12.
- Executive summary of the clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults. Arch Intern Med 1998; 158: 1855-1867.
- Krauss RM, Eckel RH, Howard B, et al. AHA Dietary Guidelines: Revision 2000. A statement for healthcare professionals from the Nutrition Committee of the American Heart Association. Stroke 2000; 31: 2751-2766.
- Clark MJ Jr, Sterrett JJ, Carson DS. Diabetes guidelines: a summary and comparison of the recommendations of the American Diabetes Association, Veterans Health Administration, and American Association of Clinical Endocrinologists. Clin Ther 2000; 22: 899-910; discussion 898.
- Esposito K, Marfella R, Ciotola M, et al. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA 2004; 292: 1440-1446.
- Tortosa A, Bes-Rastrollo M, Sanchez-Villegas A, Basterra-Gortari FJ, Nunez-Cordoba JM, Martinez-Gonzalez MA. Mediterranean diet inversely associated with the incidence of metabolic syndrome: the SUN prospective cohort. Diabetes Care 2007; 30: 2957-2959.
- Salas-Salvado J, Fernandez-Ballart J, Ros E, et al. Effect of a Mediterranean diet supplemented with nuts on metabolic syndrome status: one-year results of the PREDIMED randomized trial. Arch Intern Med 2008; 168: 2449-2458.
- Azadbakht L, Mirmiran P, Esmaillzadeh A, Azizi T, Azizi F. Beneficial effects of a Dietary Approaches to Stop Hypertension eating plan on features of the metabolic syndrome. Diabetes Care 2005; 28: 2823-2831.
- Brand-Miller J, Hayne S, Petocz P, Colagiuri S. Low-glycemic index diets in the management of diabetes: a meta-analysis of randomized controlled trials. Diabetes Care 2003; 26: 2261-2267.
- McKeown NM, Meigs JB, Liu S, Saltzman E, Wilson PW, Jacques PF. Carbohydrate nutrition, insulin resistance, and the prevalence of the metabolic syndrome in the Framingham Offspring Cohort. Diabetes Care 2004; 27: 538-546.
- Palmer M, Schaffner F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology 1990; 99: 1408-1413.
- Vajro P, Fontanella A, Perna C, Orso G, Tedesco M, De Vincenzo A. Persistent hyperaminotransferasemia resolving after weight reduction in obese children. J Pediatr 1994; 125: 239-241.
- Ueno T, Sugawara H, Sujaku K, et al. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J Hepatol 1997; 27: 103-107.
- Wang RT, Koretz RL, Yee HF Jr. Is weight reduction an effective therapy for nonalcoholic fatty liver? A systematic review. Am J Med 2003; 115: 554-559.
- Kadayifci A, Merriman R, Bass NM. Medical treatment of non-alcoholic steatohepatitis. Clin Liver Dis 2007; 11: 119140.
- Andersen T, Gluud C, Franzmann MB, Christoffersen P. Hepatic effects of dietary weight loss in morbidly obese subjects. J Hepatol 1991; 12: 224-229.
- Suzuki A, Lindor K, St Saver J, et al. Effect of changes on body weight and lifestyle in nonalcoholic fatty liver disease. J Hepatol 2005; 43: 1060-1066.
- Huang MA, Greenson JK, Chao C, et al. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol 2005; 100: 1072-1081.
- Oza N, Eguchi Y, Mizuta T, et al. A pilot trial of body weight reduction for nonalcoholic fatty liver disease with a home-based lifestyle modification intervention delivered in collaboration with interdisciplinary medical staff. J Gastroenterol 2009; 44: 1203-1208.
- Larson-Meyer DE, Newcomer BR, Heilbronn LK, et al. Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity (Silver Spring) 2008; 16: 1355-1362.
- Harrison SA, Fincke C, Helinski D, Torgerson S, Hayashi P. A pilot study of orlistat treatment in obese, non-alcoholic steatohepatitis patients. Aliment Pharmacol Ther 2004; 20: 623-628.
- Zelber-Sagi S, Kessler A, Brazowsky E, et al. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2006; 4: 639-644.
- Hussein O, Grosovski M, Schlesinger S, Szvalb S, Assy N. Orlistat reverse fatty infiltration and improves hepatic fibrosis in obese patients with nonalcoholic steatohepatitis (NASH). Dig Dis Sci 2007; 52: 2512-2519.
- Sabuncu T, Nazligul Y, Karaoglanoglu M, Ucar E, Kilic FB. The effects of sibutramine and orlistat on the ultrasonographic findings, insulin resistance and liver enzyme levels in obese patients with non-alcoholic steatohepatitis. Rom J Gastroenterol 2003; 12: 189-192.
- Kim SH, Lee YM, Jee SH, Nam CM. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11: 1116-1123.
- Padwal R, Li SK, Lau DC. Long-term pharmacotherapy for obesity and overweight. Cochrane Database Syst Rev 2004; CD004094.
- Kaya A, Aydin N, Topsever P, et al. Efficacy of sibutramine, orlistat and combination therapy on short-term weight management in obese patients. Biomed Pharmacother 2004; 58: 582-587.
- Sjostrom L, Lindroos AK, Peltonen M, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. Swedish Obese Subjects Study Scientific Group. N Engl J Med 2004; 351: 2683-2693.
- Shaffer EA. Bariatric surgery: a promising solution for nonalcoholic steatohepatitis in the very obese. J Clin Gastroenterol 2006; 40(Suppl 1): S44-S50.
- Campbell JM, Hunt TK, Karam JH, Forsham PH. Jejunoileal bypass as a treatment of morbid obesity. Arch Intern Med 1977; 137: 602-610.
- Luyckx FH, Desaive C, Thiry A, et al. Liver abnormalities in severely obese subjects: effect of drastic weight loss after gastroplasty. Int J Obes Relat Metab Disord 1998; 22: 222-226.
- Dixon JB, Bhathal PS, Hughes NR, OBrien PE. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 2004; 39: 1647-1654.
- Stratopoulos C, Papakonstantinou A, Terzis I, et al. Changes in liver histology accompanying massive weight loss after gastroplasty for morbid obesity. Obes Surg 2005; 15: 1154-1160.
- Dixon JB, Bhathal PS, OBrien PE. Weight loss and non-alcoholic fatty liver disease: falls in gamma-glutamyl transferase concentrations are associated with histologic improvement. Obes Surg 2006; 16: 1278-1286.
- Jaskiewicz K, Raczynska S, Rzepko R, Sledzinski Z. Nonalcoholic fatty liver disease treated by gastroplasty. Dig Dis Sci 2006; 51: 21-26.
- Barker KB, Palekar NA, Bowers SP, Goldberg JE, Pulcini JP, Harrison SA. Non-alcoholic steatohepatitis: effect of Roux-en-Y gastric bypass surgery. Am J Gastroenterol 2006; 101: 368-373.
- Klein S, Fontana L, Young VL, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med 2004; 350: 2549-2557.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106: 388-391.
- Eberly LE, Prineas R, Cohen JD, et al. Metabolic syndrome: risk factor distribution and 18-year mortality in the multiple risk factor intervention trial. Diabetes Care 2006; 29: 123-130.
- Bailey CJ. Biguanides and NIDDM. Diabetes Care 1992; 15: 755-772.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334: 574-579.
- Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med 1995; 333: 550-554.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2006; 29: 1963-1972.
- Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med 2000; 6: 998-1003.
- Nair S, Diehl AM, Perrillo RP. Metformin in non-alcoholic steatohepatitis (NASH): efficacy and safety - a preliminary report. Gastroenterology 2002; 122(Suppl): 4.
- Nair S, Diehl AM, Wiseman M, Farr GH Jr, Perrillo RP. Metformin in the treatment of non-alcoholic steatohepatitis: a pilot open label trial. Aliment Pharmacol Ther 2004; 20: 23-28.
- Uygun A, Kadayifci A, Isik AT, et al. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2004; 19: 537-544.
- Bugianesi E, Gentilcore E, Manini R, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol 2005; 100: 1082-1090.
- Nobili V, Manco M, Ciampalini P, et al. Metformin use in children with nonalcoholic fatty liver disease: an open-label, 24-month, observational pilot study. Clin Ther 2008; 30: 1168-1176.
- Nar A, Gedik O. The effect of metformin on leptin in obese patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Acta Diabetol 2009; 46: 113-118.
- Omer Z, Cetinkalp S, Akyildiz M, et al. Efficacy of insulin-sensitizing agents in nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 2010; 22: 18-23.
- Haukeland JW, Konopski Z, Eggesbo HB, et al. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scand J Gastroenterol 2009; 44: 853-860.
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med 2004; 351: 1106-1118.
- Da Silva Morais A, Lebrun V, Abarca-Quinones J, et al. Prevention of steatohepatitis by pioglitazone: implication of adiponectin-dependent inhibition of SREBP-1c and inflammation. J Hepatol 2009; 50: 489-500.
- Nawrocki AR, Rajala MW, Tomas E, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 2006; 281: 2654-2660.
- Fruebis J, Tsao TS, Javorschi S, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA 2001; 98: 2005-2010.
- Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 2003; 112: 91-100.
- Kamada Y, Tamura S, Kiso S, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 2003; 125: 1796-1807.
- Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 2000; 96: 1723-1732.
- Wierzbicki M, Chabowski A, Zendzian-Piotrowska M, Gorski J. Differential effects of in vivo PPAR alpha and gamma activation on fatty acid transport proteins expression and lipid content in rat liver. J Physiol Pharmacol 2009; 60: 99-106.
- Karbowska J, Kochan Z. Role of adiponectin in the regulation of carbohydrate and lipid metabolism. J Physiol Pharmacol 2006; 57(Suppl 6): 103-113.
- Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. Biochem Biophys Res Commun 2004; 315: 187-195.
- Nan YM, Fu N, Wu WJ, et al. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand J Gastroenterol 2009; 44: 358-365.
- Uto H, Nakanishi C, Ido A, et al. The peroxisome proliferator-activated receptor-gamma agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats fed a choline-deficient, l-amino acid-defined diet. Hepatol Res 2005; 32: 235-242.
- Promrat K, Lutchman G, Uwaifo GI, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004; 39: 188-196.
- Ratziu V, Giral P, Jacqueminet S, et al.; LIDO Study Group. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008; 135: 100-110.
- Idilman R, Mizrak D, Corapcioglu D, et al. Clinical trial: insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2008; 28: 200-208.
- Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008; 135: 1176-1184.
- Sanyal AJ, Mofrad PS, Contos MJ, et al. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2004; 2: 1107-1115.
- Yoneda M, Endo H, Nozaki Y, et al. Life style-related diseases of the digestive system: gene expression in nonalcoholic steatohepatitis patients and treatment strategies. J Pharmacol Sci 2007; 105: 151-156.
- Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 2003; 38: 1008-1017.
- Argo CK, Iezzoni JC, Al-Osaimi AM, Caldwell SH. Thiazolidinediones for the treatment in NASH: sustained benefit after drug discontinuation? J Clin Gastroenterol 2009; 43: 565-568.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007; 356: 2457-2471.
- Chalasani NP, Sanyal AJ, Kowdley KV, et al. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp Clin Trials 2009; 30: 88-96.
- Alberti KG, Zimmet P, Shaw J. The metabolic syndrome - a new worldwide definition. Lancet 2005; 366: 1059-1062.
- Ballantyne CM, Olsson AG, Cook TJ, Mercuri MF, Pedersen TR, Kjekshus J. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation 2001; 104: 3046-3051.
- Pyorala K, Ballantyne CM, Gumbiner B, et al. Reduction of cardiovascular events by simvastatin in nondiabetic coronary heart disease patients with and without the metabolic syndrome: subgroup analyses of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 2004; 27: 1735-1740.
- Laurin J, Lindor KD, Crippin JS, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology 1996; 23: 1464-1467.
- Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol 1999; 31: 384.
- Horlander JC, Kwo PY, Cummings OW, Koukoulis G. Atorvastatin for the treatment of NASH. Gastroenterology 2001; 120(Suppl): 2767.
- Kiyici M, Gulten M, Gurel S, et al. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol 2003; 17: 713-718.
- Gomez-Dominguez E, Gisbert JP, Moreno-Monteagudo JA, Garcia-Buey L, Moreno-Otero R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther 2006; 23: 1643-1647.
- Rallidis LS, Drakoulis CK, Parasi AS. Pravastatin in patients with nonalcoholic steatohepatitis: results of a pilot study. Atherosclerosis 2004; 174: 193-196.
- Athyros VG, Mikhailidis DP, Didangelos TP, et al. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin 2006; 22: 873-883.
- Ekstedt M, Franzen LE, Mathiesen UL, Holmqvist M, Bodemar G, Kechagias S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol 2007; 47: 135-141.
- Nozaki Y, Fujita K, Yoneda M, et al. Long-term combination therapy of ezetimibe and acarbose for non-alcoholic fatty liver disease. J Hepatol 2009; 51: 548-556.
- Cohen DE, Anania FA, Chalasani N. An assessment of statin safety by hepatologists. National Lipid Association Statin Safety Task Force Liver Expert Panel. Am J Cardiol 2006; 97: 77C-81C.
- Chalasani N, Aljadhey H, Kesterson J, Murray MD, Hall SD. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology 2004; 126: 1287-1292.
- Khorashadi S, Hasson NK, Cheung RC. Incidence of statin hepatotoxicity in patients with hepatitis C. Clin Gastroenterol Hepatol 2006; 4: 902-907.
- Lewis JH, Mortensen ME, Zweig S, Fusco MJ, Medoff JR, Belder R. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology 2007; 46: 1453-1463.
- Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology 2005; 41: 690-695.
- Riley P, Sudarshi D, Johal M, et al. Weight loss, dietary advice and statin therapy in non-alcoholic fatty liver disease: a retrospective study. Int J Clin Pract 2008; 62: 374-381.
- Georgescu EF. Angiotensin receptor blockers in the treatment of NASH/NAFLD: Could they be a first-class option? Adv Ther 2008; 25: 1141-1174.
- Yokohama S, Nakamura K, Haneda M. Clinical utility of angiotensin II receptor antagonist. Nippon Rinsho 2006; 64: 1152-1156.
- Yokohama S, Tokusashi Y, Nakamura K, et al. Inhibitory effect of angiotensin II receptor antagonist on hepatic stellate cell activation in nonalcoholic steatohepatitis. World J Gastroenterol 2006; 12: 322-326.
- Yokohama S, Yoneda M, Haneda M, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004; 40: 1222-1225.
- Enjoji M, Kotoh K, Kato M, et al. Therapeutic effect of ARBs on insulin resistance and liver injury in patients with NAFLD and chronic hepatitis C: a pilot study. Int J Mol Med 2008; 22: 521-527.
- Georgescu EF, Ionescu R, Niculescu M, Mogoanta L, Vancica L. Angiotensin-receptor blockers as therapy for mild-to-moderate hypertension-associated non-alcoholic steatohepatitis. World J Gastroenterol 2009; 15: 942-954.
- Rosselli MS, Burgueno AL, Carabelli J, Schuman M, Pirola CJ, Sookoian S. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis 2009; 206: 119-126.
- Kudo H, Yata Y, Takahara T, et al. Telmisartan attenuates progression of steatohepatitis in mice: role of hepatic macrophage infiltration and effects on adipose tissue. Liver Int 2009; 29: 988-996.
- Chang CY, Argo CK, Al-Osaimi AM, Caldwell SH. Therapy of NAFLD: antioxidants and cytoprotective agents. J Clin Gastroenterol 2006; 40(Suppl 1): S51-S60.
- Guma C, Viola L, Thome M, Galdame O, Alvarez E. Ursodeoxycholic acid in the treatment of non-alcoholic steatohepatitis: results of prospective clinical controlled trial. Hepatology 1997; 26(Suppl): 1036.
- Ceriani R, Bunati S, Morini L, Sacchi E, Colombo G. Effect of ursodeoxycholic acid plus diet in patients with non-alcoholic steatohepatitis. Hepatology 1998; 28(Suppl): 894.
- Holoman J, Glasa J, Kassar J, et al. Serum markers of liver fibrosis in patients with non-alcoholic steatohepatitis (NASH): correlation to liver morphology and effect of therapy. J Hepatol 2000; 32(Suppl): 210.
- Lindor KD, Kowdley KV, Heathcote EJ, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology 2004; 39: 770-778.
- Dufour JF, Oneta CM, Gonvers JJ, et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin E in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2006; 4: 1537-1543.
- Balmer ML, Siegrist K, Zimmermann A, Dufour JF. Effects of ursodeoxycholic acid in combination with vitamin E on adipokines and apoptosis in patients with nonalcoholic steatohepatitis. Liver Int 2009; 29: 1184-1188.
- Czaja MJ, Weiner FR, Flanders KC, et al. in vitro and in vivo association of transforming growth factor-b1 with hepatic fibrosis. J Cell Biol 1989; 108: 2477-2482.
- Eghbali-Fatourechi G, Sieck GC, Prakash YS, Maercklein P, Gores GJ, Fitzpatrick LA. Type I procollagen production and cell proliferation is mediated by transforming growth factor-b in a model of hepatic fibrosis. Endocrinology 1996; 137: 1894-1903.
- Sanderson N, Factor V, Nagy P, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA 1995; 92: 25722576.
- Parola M, Muraca R, Dianzani I, et al. Vitamin E dietary supplementation inhibits tissue growth factor beta-1 gene expression in the rat liver. FEBS Lett 1992; 308: 267-270.
- Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther 2001; 15: 1667-1672.
- Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. J Pediatr 2000; 136: 734-738.
- Vajro P, Mandato C, Franzese A, et al. Vitamin E treatment in pediatric obesity-related liver disease: a randomized study. J Pediatr Gastroenterol Nutr 2004; 38: 48-55.
- Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003; 38: 413-419.
- Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2003; 98: 2485-2490.
- Nobili V, Manco M, Devito R, et al. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: a randomized, controlled trial. Hepatology 2008; 48: 119-128.
- Takitani K, Miyazaki H, Yoden A, Tamai H. Childrens toxicology from bench to bed-liver injury (2): mechanism of antioxidant therapy for nonalcoholic fatty liver disease. J Toxicol Sci 2009; 34(Suppl 2): SP223-SP228.
- Lavine JE, Schwimmer JB, Molleston JP, et al. Treatment of nonalcoholic fatty liver disease in children: TONIC trial design. Contemp Clin Trials 2009.
- Reddy JK. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal
ß-oxidation, PPAR
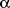 ,
and steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2001; 281:
G1333-G1339.
,
and steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2001; 281:
G1333-G1339.
- Araya J, Rodrigo R, Videla LA, et al. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004; 106: 635-643.
- Clarke SD. Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am J Physiol Gastrointest Liver Physiol 2001; 281: G865G869.
- Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARa agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004; 39: 1286-1296.
- Kiec-Wilk B, Dembinska-Kiec A, Olszanecka A, Bodzioch M, Kawecka-Jaszcz K. The selected pathophysiological aspects of PPARs activation. J Physiol Pharmacol 2005; 56: 149-162.
- Levy JR, Clore JN, Stevens W. Dietary n-3 polyunsaturated fatty acids decrease hepatic triglycerides in Fisher 344 rats. Hepatology 2004; 39: 608-616.
- Storlien LH, Kraegen EW, Chisholm DJ, Ford GL, Bruce DG, Pascoe WS. Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 1987; 237: 885-888.
- Capanni M, Calella F, Biagini MR, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther 2006; 23: 1143-1151.
- Zhu FS, Liu S, Chen XM, Huang ZG, Zhang DW. Effects of n-3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J Gastroenterol 2008; 14: 6395-6400.
- Satapathy SK, Garg S, Chauhan R, et al. Beneficial effects of tumor necrosis factor-alpha inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2004; 99: 1946-1952.
- Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol 2004; 99: 2365-2368.
- Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol 2007; 22: 634-638.
- Lee YM, Sutedja DS, Wai CT, et al. A randomized controlled pilot study of Pentoxifylline in patients with non-alcoholic steatohepatitis (NASH). Hepatol Int 2008; 2: 196-201.
- de la Lastra CA, Villegas I. Resveratrol as an anti-inflamatory and anti-aging agent: mechanisms an clinical implications. Mol Nutr Food Res 2005; 49: 405-430.
- Bujanda L, Hijona E, Larzabal M, et al. Resveratrol inhibits nonalcoholic fatty liver disease in rats. BMC Gastroenterol 2008; 8: 40-45.
- Culleton BF, Larson MG, Kannel WB, Levy D. Serum uric acid and risk for cardiovascular disease and death: The Framingham Heart Study. Ann Intern Med 1999; 131: 7-13.
- Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008; 359: 1811-1821.
- Suzuki I, Yamauchi T, Onuma M, Nozaki S. Allopurinol, an inhibitor of uric acid synthesis-can it be used for the treatment of metabolic syndrome and related disorders? Drugs Today (Barc) 2009; 45: 363-378.
- Solga SF, Diehl AM. Non-alcoholic fatty liver disease: lumen-liver interactions and possible role for probiotics. J Hepatol 2003; 38: 681-687.
- Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol 2005; 39: 540-543.
- Velayudham A, Dolganiuc A, Ellis M, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009; 49: 989-997.
- Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006; 43: 173-181.
- Tushuizen ME, Bunck MC, Pouwels PJ, van Waesberghe JH, Diamant M, Heine RJ. Incretin mimetics as a novel therapeutic option for hepatic steatosis. Liver Int 2006; 26: 1015-1017.