Proteinase-activated receptors (PARs), a family
of G-protein-coupled seven-transmembrane receptors, are activated by proteolytic
cleavage of the N-terminal domain, and subsequent binding of the newly appeared
N-terminus, a tethered ligand, to the receptor itself (1, 2). PAR1, PAR2 and
PAR4, but not PAR3, can also be activated by synthetic PAR-activating peptides
based on the tethered ligand sequences (2, 3). PARs, upon activation, induce
various physiological/pathophysiological responses, including platelet aggregation,
pro- or anti-inflammatory responses, gastric mucosal protection, exocrine secretion,
accelerating proliferation, processing of pain signals, regulation of smooth
muscle motility,
etc. (2, 4, 5). Interestingly, PARs are activated by
not only endogenous enzymes,
e.g. thrombin, trypsin, tryptase, coagulation
factors VIIa and Xa, plasmin, kallikrein,
etc., but also exogenous serine
proteinases, such as gingipains-R derived from
Porphyromonas gingivalis,
the pathogenesis bacteria of periodontitis, Der p3 and Der p9, house dust mite
allergens, and cockroach proteinases (3, 6).
Helicobacter pylori (
H. pylori), a microaerophilic Gram-negative bacterium, colonizes the human stomach. Infection with
H. pylori is a well-known risk factor for gastroduodenal ulcers and for gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT) lymphoma (7-9).
H. pylori possesses various pathogenicity factors including CagA, VacA, peptidoglycan, lipopolysaccharide (LPS), urease and secretory proteinases, which trigger various signals for inflammation, immunity, proliferation, survival, and so on (9, 10). The secretory proteinases include HtrA and urokinase-type plasminogen activator (u-PA) (11, 12), Interestingly, there is evidence that
H. pylori-derived proteinases cause interleukin-8 (IL-8) production through activation of PAR2 in gastric epithelial MKN45 cells (13, 14). On the contrary, it has also been reported that PAR1 protects the host against severe
H. pylori-induced gastritis
via suppression of production of macrophage-inflammatory protein (MIP)-2, the mouse IL-8 functional homologue, in mice (15). Another paper shows that PAR2 attenuates
H. pylori-induced cell death and DNA fragmentation in gastric AGS cells (16). These reports strongly suggest that PARs may contribute to the
H. pylori-related events in the gastric mucosa, although it is open to question whether PARs suppress or aggravate the
H. pylori-induced pathological responses. In the present study, to examine if the
H. pylori actually possesses proteinases that can activate PARs, we prepared the supernatant of
H. pylori homogenate (
H. pylori extracts), and evaluated the biological activity of the components in gastrointestinal and other cell lines that functionally express PAR1, PAR2 or PAR4 (17-19), and in rat platelets that express PAR3 and PAR4 (20, 21).
MATERIAL AND METHODS
Materials
The PAR1-activating peptide (PAR1-AP) Thr-Phe-Leu-Leu-Arg-amide (TFLLR-NH
2),
the PAR2-AP Ser-Leu-Ile-Gly-Arg-Leu-amide (SLIGRL-NH
2)
and the PAR4-AP Ala-Tyr-Pro-Gly-Lys-Phe-amide (AYPGKF-NH
2)
were synthesized and purified by high-performance liquid chromatography (HPLC),
and the concentration and purity were determined by HPLC or mass spectrometry.
Nafamostat mesilate was a gift from Torii Pharmaceutical Company (Tokyo, Japan).
Polymyxin B sulfate, IRAK-1/4-inhibitor I (IRAK-1/4-I), genistein, pyrrolidine
dithiocarbamate (PDTC), U0126 and dimethyl sulfoxide (DMSO) were purchased from
Sigma-Aldrich (St. Louis, MO), SP600125, SB203580 and BAY11-7082 were from Calbiochem
(Darmstadt, Germany), and LY294002 was from Tocris Bioscience (Ellisville, MO).
PAR-activating peptides were dissolved in saline. Polymyxin B and PDTC were
dissolved in distilled water, and the other chemicals were in DMSO.
Culture of H. pylori and preparation of the extracts
H. pylori (ATCC 43504, Rockville, MD) were cultured on Trypticase soy agar (Becton, Dickinson and Company, Sparks, MD) with 10% defibrinated sheep blood (Japan Lamb, Hiroshima, Japan) at 37°C under microaerophilic conditions, and then harvested in the ice-cold sterilized phosphate-buffered saline (PBS). The suspension of
H. pylori was sonicated, and then centrifuged. The supernatant was collected and filtered through a filter with 0.22 µm pore. The filterate was used as the
H. pylori extracts. The protein concentration of the extracts was measured with the Bradford protein assay method (Bio-Rad Laboratories, Tokyo, Japan).
Cell culture
Normal rat gastric epithelial mucosal RGM1 cells were cultured in DMEM F-12 Ham (Sigma-Aldrich) supplemented with 20% fetal calf serum (FCS) (Thermo, Melbourne, Australia) and 50 mg/L kanamycin (Meiji Seika Kaisha, Ltd., Tokyo, Japan). Human lung adenocarcinoma A549 cells and human colorectal adenocarcinoma HCT-15 cells were cultured in DMEM supplied with 10% FCS and 50 mg/L kanamycin.
Assay of PGE2 and IL-8 release
RGM1, A549 and HCT-15 cells (1.52×10
5 cells/well
of 6-well plate) were cultured in the above-mentioned FCS-containing medium
for 24 hours, and then, cultured in the FCS-free medium overnight. One hour
after the medium was refreshed, the cells were stimulated with the
H. pylori
extracts or the PAR-APs. Inhibitors were added 30 min before the stimulation.
Small volume samples were collected from the culture medium at appropriate points
in time. The concentration of PGE
2 and IL-8
in the collected samples was assayed using the PGE
2
EIA kit (Cayman Chem., Ann Arbor, MI, USA) and IL-8 ELISA kit (Amersham Biosciences,
Buckinghamshire, UK), respectively.
Western blotting
A549 cells were cultured, and then stimulated with the PAR2-AP SLIGRL-NH
2
or the
H. pylori extracts, as mentioned above. After the stimulation,
the cells were lysed in sodium dodecyl sulfate (SDS) buffer (2% SDS, 62.5 mM
Tris-HCl, and 10% glycerol, pH 6.8). Protein samples (30 µg for COX-2, 10 µg
for others) were separated by electrophoresis on 12.5% SDS-polyacrylamide gel
(Wako Pure Chemicals, Osaka, Japan), and transferred onto polyvinylidene difluoride
membrane (Millipore, Bedford, MA). After blocking the membrane with a blocking
buffer (5% skim milk, 137 mM NaCl, 0.1% Tween-20 and 20 mM Tris-HCl, pH 7.6),
the membrane was incubated with primary antibodies overnight at 4°C, and then
washed and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-goat
antibodies (Cell Signaling Technol.). The primary antibodies used in the present
study were: the anti-COX-2 and anti-GAPDH antibodies from Santa Cruz Biotech.
(Santa Cruz, CA), anti-p44/42 MAP kinase and anti-phospho-p44/42 MAP kinase
antibodies from Cell Signaling Technol. (Beverly, MA). Positive bands were developed
by enhanced chemiluminescence detention Western blotting detection reagent (Amersham
Biosciences).
Experimental animals
Male Wistar rats weighing 150200 g were purchased from Japan SLC Inc. (Shizuoka, Japan) and used at the body weight of 300400 g. All animals were used with approval by the Committee for the Care and Use of Laboratory Animals at Kinki University, and all procedures employed in the present study were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 80-23, revised 1996).
Platelet aggregation
Under anesthesia with urethane at 1.5 g/kg, blood was collected from the abdominal
aorta of rats into a plastic syringe containing one-tenth volume of 3.8% sodium
citrate (Fuso Pharmaceutical Industries Ltd., Osaka, Japan). Platelet-rich plasma
(PRP) and washed platelets at 4×10
5 platelets/µL
were prepared from the blood at room temperature as described previously (22,
23). Platelet aggregation was monitored as an increase in light transmission
at 37°C a platelet aggregometer (SSR Engineering Co. Ltd., Tokyo, Japan).
Statistical analysis
All data are represented as mean ±S.E.M. Statistical analysis was performed by Tukey's test. Significance was set at P<0.05 level.
RESULTS
Determination of the activity of H. pylori extracts in RGM1 cells expressing PAR1 and in A549 cells expressing PAR2
In normal rat gastric mucosal epithelial RGM1 cells that abundantly express
PAR1, activation of PAR1 with the PAR1-AP TFLLR-NH
2
caused PGE
2 release (
Fig. 1A), as reported
previously (18). On the other hand,
H. pylori extracts did not induce
PGE
2 release in RGM1 cells (
Fig. 1A).
 |
Fig. 1. Activity of H.
pylori extracts in the PAR1-expressing RGM1 cells and PAR2-expressing
A549 cells. (A and B) RGM1 (A) and A549 (B) cells were stimulated with
H. pylori extracts, the PAR1-activating peptide (AP), TFLLR-NH2,
or the PAR2-AP, SLIGRL-NH2, for 24 hours
(A) or 12 hours (B), and the released PGE2 was quantified. **P<0.01
vs. vehicle. Data show the mean ±S.E.M. from 4–8 experiments.
(C and D) COX-2 (C) and phosphorylated ERK1/2 (p-ERK1/2) (D) were detected
by Western blotting in A549 cells stimulated with SLIGRL-NH2
at 100 µM and the H. pylori extract at 100 µg protein/ml. |
In human lung adenocarcinoma A549 cells that abundantly express PAR2,
H.
pylori extracts significantly increased PGE
2
release (
Fig. 1B), and caused upregulation of COX-2 (
Fig. 1C)
and phosphorylation of ERK (
Fig. 1D), as the PAR2-AP, SLIGRL-NH
2,
did (
Fig. 1B-1D). The PGE
2 release induced
by
H. pylori extracts was resistant to nafamostat mesilate, an inhibitor
of proteinases with a broad spectrum (
Fig. 2A), and exposure to boiling
(
Fig. 2B). On the other hand, polymyxin B, an LPS inhibitor, IRAK-1/4
inhibitor-I, and PDTC, an NF-
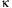
B
inhibitor, significantly suppressed the effect of the
H. pylori extracts
(
Fig. 2C-2E).
 |
Fig. 2. Characterization of
the PGE2 release caused by the H.
pylori extracts in A549 cells. (A and B) nafamostat mesilate, a proteinase
inhibitor, and boiling of the H. pylori extracts did not affect
the evoked PGE2 release. (C, D and E)
Effects of polymyxin B, an inhibitor of LPS, IRAK-1/4 inhibitor I (IRAK-1/4-I),
or PDTC, an NF-B
inhibitor on the PGE2 release by the
H. pylori extracts. Polymyxin B was incubated with H. pylori
extracts for 30 min, and then, the mixture was added to the cells. IRAK-1/4-I
or PDTC was added 30 min before stimulation with H. pylori extracts.
* P<0.05, ** P<0.01 vs. vehicle + vehicle (V+V). † P<0.05,
†† P<0.01 vs. V+H. pylori extracts. Data show the
mean ±S.E.M. from 4–7 experiments. |
Evaluation of the activity of H. pylori extracts in HCT-15 cells known to express PAR4
In human colorectal adenocarcinoma HCT-15 cells that abundantly express PAR2,
not only the PAR2-AP, SLIGRL-NH
2, but also the
PAR4-AP, AYPGKF-NH
2, significantly caused IL-8
release (
Fig. 3A), suggesting that PAR4 is also expressed in HCT-15 cells.
H. pylori extracts induced IL-8 release in HCT-15 cells (
Fig. 3A),
an effect being partially, but significantly suppressed by nafamostat mesilate,
a proteinase inhibitor, or boiling of the extracts (
Fig. 3B). The effect
of the
H. pylori extracts was strongly suppressed by IRAK-1/4 inhibitor-I
(
Fig. 3D), but not by inhibitors of LPS (
Fig. 3C) or NF-
B
(
Fig. 3E). On the other hand, inhibitors of MEK (U0126), JNK (SP600125)
and p38 MAP kinase (SB203580) significantly suppressed the activity of the
H.
pylori extracts (
Fig. 3F).
 |
| Fig.
3. Characterization of IL-8 release caused by H. pylori extracts
in HCT-15 cells. (A) Stimulation with the PAR2-AP, SLIGRL-NH2,
PAR4-AP, AYPGKF-NH2, and H. pylori
extracts, but not vehicle (V), caused IL-8 release. (B) The IL-8 release
caused by H. pylori extracts was partially suppressed by nafamostat
mesilate, a proteinase inhibitor, and the exposure to boiling. (C, D,
E and F) Effects of an LPS inhibitor (polymyxin B) (C), IRAK-1/4 inhibitor
I (IRAK-1/4-I) (D), and inhibitors of NF-B
(PDTC and BAY 11-7082) (E) and MEK (U0126), JNK (SP600125), or p38 MAP
kinase (SB203580) (F) on the IL-8 release caused by H. pylori extracts.
** P<0.01 vs. vehicle + vehicle (V+V). † P<0.05, ††
P<0.01 vs. V+H. pylori extract. Data show the mean ±S.E.M.
from 4–7 experiments. |
Assessment of the activity of H. pylori extracts in rat platelets that express PAR4 and PAR3
To determine whether
H. pylori extracts include PAR4-activating proteinases,
we used rat platelets, since rat platelets express PAR4 and PAR3 that mediate
the thrombin-induced platelet aggregation (21). In rat platelet-rich plasma
(PRP),
H. pylori extracts caused aggregation, as the PAR4-AP, AYPGKF-NH
2,
did (
Fig. 4A). However, nafamostat mesilate, a proteinase inhibitor,
did not suppress the activity of the
H. pylori extracts in PRP (
Fig.
4A). Further, in the washed platelets that responded to AYPGKF-NH
2,
the
H. pylori extracts failed to cause aggregation (
Fig. 4B).
 |
Fig. 4. Activity of H.
pylori extracts in rat platelets. (A) Platelet aggregation caused
by H. pylori extracts in platelet-rich plasma (PRP). (B) Lack of
the activity of H. pylori extracts in the washed platelets. Platelets
were stimulated with the PAR4-AP, AYPGKF-NH2,
at 300 µM or H. pylori extracts at 100 µg protein/mL
in the absence or presence of nafamostat mesilate, a proteinase inhibitor. |
DISCUSSION
In the present study, we first found that the PAR1-AP but not
H. pylori
extracts caused PGE
2 release in RGM1 cells that
express PAR1 (18, 24), suggesting that the
H. pylori extracts do not
contain PAR1-activating proteinases.
Next, we showed that
H. pylori extracts, like the PAR2-AP, induced PGE
2
release in A549 cells known to abundantly express PAR2 (17, 25, 26). However,
this effect of the
H. pylori extracts is not considered to be mediated
by proteinases, since it was resistant to nafamostat mesilate or exposure to
boiling. LPS, one of heat-resistant pathogenetic factors of
H. pylori,
might be responsible for the PGE
2 release induced
by
H. pylori extracts, considering the findings that the effect of the
H. pylori extracts was suppressed by polymixin B, an inhibitor of LPS,
and by inhibitors of IRAK-1/4 and NF-
B,
downstream signals for toll-like receptor (TLR) 2, a receptor of
H. pylori-derived
LPS (9, 27, 28). The pathological roles of
H. pylori LPS have been described
in human peripheral blood mononuclear leukocytes (29), although it is not highly
toxic.
Our group has reported that human colorectal adenocarcinoma HCT-15 cells release
IL-8 in response to the PAR2-AP SLIGRL-NH
2,
but not PAR1-AP TFLLR-NH
2 (19). In the present
study, expression of functional PAR4 in addition to PAR2 in HCT-15 cells was
demonstrated by the effectiveness of the PAR4-AP (see
Fig. 3A). Our present
findings that
H. pylori extracts caused release of IL-8 in HCT-15 cells,
an effect partially suppressed by nafamostat mesilate and exposure to boiling,
could suggest the possibility of the presence of PAR4-activating proteinases
in
H. pylori extracts, which lack PAR2-activating proteinases, as shown
in A549 cells (see
Fig. 2A, 2B). The proteinase-independent and heat-resistant
portion of the IL-8 release caused by
H. pylori extracts in HCT-15 cells
does not appear to involve LPS, since it was resistant to polymyxin B. The inhibition
experiments suggest the involvement of IRAK1/4, MEK/ERK1/2, JNK and p38 MAP
kinase, but not NF-
B,
although the detailed signaling mechanisms are open to question. It has been
reported that
H. pylori increases H
2O
2
release
via activation of NADPH oxidase, followed by activation of the
Jak1/Stat3 pathway and RANTES release in gastric epithelial AGS cells (30).
Since IL-8 release is enhanced by the reactive oxygen species (ROS) produced
by NADPH oxidase (31), it is of interest to determine the involvement of the
NADPH oxidase/ROS/Jak1/Stat3 pathway in the
H. pylori extract-caused
IL-8 release in HCT-15 cells.
Interestingly,
H. pylori extracts caused aggregation of rat platelets
in PRP, which express PAR4 and PAR3 (21). This effect of
H. pylori extracts
was resistant to nafamostat mesilate, being inconsistent with the nafamostat
mesilate-sensitive IL-8 release by the
H. pylori extracts in HCT-15 cells.
Thus,
H. pylori does not contain PAR4- or PAR3-activating proteinases,
considering the finding that the
H. pylori extracts did not cause aggregation
in washed platelets, where the PAR4-AP (see
Fig. 4B) and thrombin, an
activator of PAR1, PAR3 and PAR4 (data not shown), are capable of causing aggregation.
Our data also suggest that the
H. pylori extracts require unknown plasma
co-factor(s) in causing platelet aggregation. It has been reported that some
strains of
H. pylori induce human platelet aggregation through their
binding to von Willebrand factor (vWF) followed by interaction with glycoprotein
Ib (32). Similar mechanisms might be involved in the
H. pylori extracts-induced
aggregation in rat PRP.
In conclusion, our study demonstrates that
H. pylori extracts mimic some of the actions of the PAR2-AP and/or PAR4-AP in A549 and HCT-15 cells or rat platelets, whereas any proteinases capable of activating PAR1, PAR2 and PAR4 are not involved in the activity of the
H. pylori components.
Acknowledgements:
This research was supported in part by Grant-in-Aid for Scientific Research
from Japan Society for the Promotion of Science, 2008-2010, and by 'Antiaging
Center Project' for Private universities from Ministry of Education, Culture,
Sports, Science and Technology, 2008-2012.
This work in its preliminary from has been presented during 7th International
Symposium on Cell/Tissue Injury and Cytoprotection Organoprotection, Sepember
9-11, 2012, Honolulu, Hawaii organized by Prof. K. Takeuchi (Kyoto, Japan) and
Prof. H. Matsui (Tsukuba, Japan).
Conflict of interests: None declared.
REFERENCES
- Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol 2010; 160: 191-203.
- Steinhoff M, Buddenkotte J, Shpacovitch V, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev 2005; 26: 1-43.
- Sekiguchi F, Kawabata A. Protease-activated receptors (PARs) as therapeutic targets: development of agonists/antagonists and modulation of gastrointestinal functions. Drug Des Rev-Online 2004; 1: 287-296.
- Sekiguchi F, Takaoka K, Kawabata A. Proteinase-activated receptors in the gastrointestinal system: a functional linkage to prostanoids. Inflammopharmacology 2007; 15: 246-254.
- Kawabata A, Matsunami M, Sekiguchi F. Gastrointestinal roles for proteinase-activated receptors in health and disease. Br J Pharmacol 2008; 153: S230-S240.
- Kawabata A. PAR-2: structure, function and relevance to human diseases of the gastric mucosa. Expert Rev Mol Med 2002; 2002: 1-17.
- Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med 2007; 13: 470-476.
- Matsuda K, Yamauchi K, Matsumoto T, Sano K, Yamaoka Y, Ota H. Quantitative analysis of the effect of Helicobacter pylori on the expressions of SOX2, CDX2, MUC2, MUC5AC, MUC6, TFF1, TFF2, and TFF3 mRNAs in human gastric carcinoma cells. Scand J Gastroenterol 2008; 43: 25-33.
- Backert S, Clyne M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2011; 16(Suppl 1): 19-25.
- Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007; 133: 288-308.
- Bumann D, Aksu S, Wendland M, et al. Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect Immun 2002; 70: 3396-3403.
- Beyer BC, Heiss MM, Simon EH, et al. Urokinase system expression in gastric carcinoma: prognostic impact in an independent patient series and first evidence of predictive value in preoperative biopsy and intestinal metaplasia specimens. Cancer 2006; 106: 1026-1035.
- Kajikawa H, Yoshida N, Katada K, et al. Helicobacter pylori activates gastric epithelial cells to produce interleukin-8 via protease-activated receptor 2. Digestion 2007; 76: 248-255.
- Yoshida N, Yoshikawa T. Basic and translational research on proteinase-activated receptors: implication of proteinase/proteinase-activated receptor in gastrointestinal inflammation. J Pharmacol Sci 2008; 108: 415-421.
- Wee JL, Chionh YT, Ng GZ, et al. Protease-activated receptor-1 down-regulates the murine inflammatory and humoral response to Helicobacter pylori. Gastroenterology 2010; 138: 573-582.
- Lim JW, Kim H. Role of protease-activated receptor-2 on cell death and DNA fragmentation in Helicobacter pylori-infected gastric epithelial cells. J Transl Med 2010; 8: 85.
- Kawao N, Nagataki M, Nagasawa K, et al. Signal transduction for
proteinase-activated receptor-2-triggered prostaglandin E2
formation in human lung epithelial cells. J Pharmacol Exp Ther 2005; 315:
576-589.
- Sekiguchi F, Saito S, Takaoka K, et al. Mechanisms for prostaglandin
E2 formation caused by proteinase-activated
receptor-1 activation in rat gastric mucosal epithelial cells. Biochem Pharmacol
2007; 73: 103-114.
- Tanaka Y, Sekiguchi F, Hong H, Kawabata A. PAR2 triggers IL-8 release via MEK/ERK and PI3-kinase/Akt pathways in GI epithelial cells. Biochem Biophys Res Commun 2008; 377: 622-626.
- Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000; 404: 609-613.
- Hollenberg MD, Saifeddine M. Proteinase-activated receptor 4 (PAR4): activation and inhibition of rat platelet aggregation by PAR4-derived peptides. Can J Physiol Pharmacol 2001; 79: 439-442.
- Hata T, Kawabata A, Itoh E. Platelet hypoaggregability in rats exposed to SART stress (repeated cold stress). Thromb Res 1992; 65: 617-629.
- Kawabata A, Hata T. Characterization of platelet hypofunctions in rats under SART stress (repeated cold stress). Thromb Res 1993; 69: 197-207.
- Sekiguchi F, Ohi A, Maeda Y, et al. Delayed production of arachidonic acid contributes to the delay of proteinase-activated receptor-1 (PAR1)-triggered prostaglandin E2 release in rat gastric epithelial RGM1 cells. J Cell Biochem 2011; 112: 909-915.
- Moriyuki K, Nagataki M, Sekiguchi F, Nishikawa H, Kawabata A. Signal transduction for formation/release of interleukin-8 caused by a PAR2-activating peptide in human lung epithelial cells. Regul Pept 2008; 145: 42-48.
- Nagataki M, Moriyuki K, Sekiguchi F, Kawabata A. Evidence that PAR2-triggered
prostaglandin E2 (PGE2)
formation involves the ERK-cytosolic phospholipase A2-COX-1-microsomal PGE
synthase-1 cascade in human lung epithelial cells. Cell Biochem Funct 2008;
26: 279-282.
- Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 2007; 51: 140-148.
- Smith SM, Moran AP, Duggan SP, et al. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol 2011; 186: 2462-2471.
- Grebowska A, Moran AP, Bielanski W, et al. Helicobacter pylori lipopolysaccharide activity in human peripheral blood mononuclear leukocyte cultures. J Physiol Pharmacol 2010; 61: 437-442.
- Cha B, Lim JW, Kim KH, Kim H. 15-deoxy-D12,14-prostaglandin J2 suppresses RANTES expression by inhibiting NADPH oxidase activation in Helicobacter pylori-infected gastric epithelial cells. J Physiol Pharmacol 2011; 62: 167-174.
- Miyoshi T, Yamashita K, Arai T, Yamamoto K, Mizugishi K, Uchiyama T. The role of endothelial interleukin-8/NADPH oxidase 1 axis in sepsis. Immunology 2010; 131: 331-339.
- Byrne MF, Kerrigan SW, Corcoran PA, et al. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003; 124: 1846-1854.