THE NON-NEURONAL HEART’S ACETYLCHOLINE IN HEALTH AND DISEASE
INTRODUCTION
Acetylcholine (ACh) mostly regarded as a transmitter in central and peripheral nervous system is, however, synthesized and released also in the cells and tissues devoid of any innervation like trophoblast of the placenta, renal tubuli, epithelial cells of airways and alimentary tract, and even some bacteria (1, 2). This review describes convincing evidence cumulating for almost one century, which shows that ACh is synthesized and released also by the cardiac myocytes. The role of the heart’s non-neuronal ACh in the heart function in health and disease is also discussed.
The heart is innervated by both vagus nerves (VNs). Their efferent preganglionic fibres form synapses with the ganglia situated in the atria, mostly in the vicinity of the sino-atrial node. The postganglionic fibres innervate richly the sino-atrial node, the atrial muscle, and atrio-ventricular node. In contrast to the rich sympathetic innervation, the VNs fibres in the ventricles are sparse, mostly limited to the conducting system, although some parasympathetic fibres are found also in the ventricular working muscle (3, 4). ACh acts as a transmitter released by both the pre- and postganglionic parasympathetic fibres.
The ACh released from the postganglionic parasympathetic fibres binds to its receptors. The M2 acetylcholine receptor (M2AChR) is the main subtype musacrinic receptor in the heart, however, M3 receptors may also play a role in the control of the heart’s function (2). ACh binding to the M2AChR results in: (1) decrease in the heart rate by inhibition of the slow diastolic depolarization of pacemaker cells of the sino-atrial node, (2) shortening of the action potentials of the atrial myocytes and thus decreasing amplitude of their contraction, (3) slowing the conduction in the sino-atrial and atrio-ventricular nodes and (4) slight decrease of the amplitude of contractions of the ventricular myocytes.
In the cholinergic neurons ACh is synthetsized by choline acetyltransferase (ChAT) from free choline and Acetyl-CoA (5). The synthesized ACh is transported into the cytoplasmic vesicles by vesicular ACh transporter (VAChT) (6). The vesicles move to and fuse with the presynaptic membrane. The released ACh binds to its receptors on the cardiomyocyte membranes and then is rapidly hydrolyzed by acetylcholine esterase (AChE). The free choline is transported back into the cholinergic fiber by the high-affinity choline transporter (CHT-1) (7) (Fig. 1).
NON-NEURONAL ACETYLCHOLINE IN THE HEART
The seminal work of Bulbring and Burn (8) initiated investigations on the presence and role of non-neuronal ACh in the heart. They investigated the spontaneous contractile activity and synthesis of ACh of the isolated rabbit atria. Freshly isolated atria were beating vigorously and synthesized considerable amounts of ACh. As could be expected, ACh added to the bath inhibited the beating. After many hours after isolation spontaneous beating of the atria stopped, and their synthesis of ACh was largely depressed. Then, ACh added to the bath stimulated spontaneous beating and restored the power of the ACh synthesis by the atria to the level comparable with that of freshly isolated ones. The authors put forward the view that “the activity of the auricular muscle and the synthesis of ACh are inseparatedly linked”.
It was not clear what was the source of endogenous ACh in Bulbring and Burn (8) experiments. It might be the cholinergic parasympathetic fibres richly innervating the atria, the atrial muscle or both. The following works of this group aimed to check whether the heart may produce ACh independently of its innervation.
Briscoe and Burn (9) found that ACh was released into the closed circuit perfusate of the isolated, spontaneously beating rabbit heart. Blocking of AChE resulted in slowing of the heart rate due to ACh accumulation in the perfusate. The effect was inhibited by atropine, the blocker of muscarinic receptors. In these experiments ACh was released from the isolated, spontaneously beating hearts, in which the VNs fibres should have not been active. Day (10) confirmed these results in spontaneously beating hearts and found that stimulation of the ventricles at the rate four times higher then spontaneous one strongly increased the ACh release to the perfusate. Removal of the atria richly innervated by cholinergic fibres reduced ACh release by one third only. Spontaneous activity of the sparse cholinergic fibres in the ventricles deprived of their ganglia removed with the atria is highly unlikely. Thus, the above referred experiments suggested that ACh released from the isolated hearts was mostly of non-neuronal origin and that release was related to the heart contractile activity.
The hypothesis of non-neuronal synthesis of ACh in the heart has been further supported in our (11) experiments with the use of hemicholinium No 3 (HC3) developed by Schueler (12). HC3 blocks CHT-1 precluding transport of choline into the cell. This results in inhibition of cellular ACh synthesis due to the lack of one of the substrates (Fig. 1). Therefore, HC3 should result in ACh depletion in the cells synthesizing and releasing of ACh. The main series of experiments were performed in spontaneously beating rabbit atria isolated with their both VNs attached. In the control experiments both nerves were stimulated supramaximally for 1 min every 4 min for 1 – 2 hours. Each stimulation resulted in complete inhibition of the atrial contractions followed by recovery. At the end of experiment the ACh content in these atria did not differ from that in the atria of which the VNs were not stimulated. These experiments show that ACh released from the nerve endings upon the VNs stimulation was constantly resynthesized. Addition of HC3 to the bath resulted in gradual decrease of the atrial response to VNs stimulation until it completely vanished within ~ 40min, showing that ACh of the parasympathetic nerve endings has been depleted (checked by the method described in (13)). The ACh content of these atria decreased by more then 50%. This result showed that resynthesis of large fraction of the ACh released from the atria in these experiments has been inhibited by HC3 and raised the question of its origin: nerve endings, myocytes or both. The following experiments provided the answer. Incubation of the beating atria with HC3 for 1 hour without stimulation of VNs also resulted in the drop of their ACh content which was only slightly less then that in the atria of which VNs were stimulated. Importantly, at the end of incubation the VNs were fully active in these atria which showed that ACh of their endings has not been depleted. This result clearly showed that the large ACh fraction depleted from the atria the VNs of which were not stimulated must have been of non-neuronal origin.
The following stimulation of VNs in these atria till the exhaustion of the response resulted in only negligible further drop in ACh content which showed that the contribution of the neuronal ACh to its total atrial content is very small. Further, it was found that HC3 did not deplete ACh from the isolated quiescent left atrial appendix unless it was stimulated at the rate comparable to the spontaneous one.
Interestingly, HC3 did not affect the in situ atrial (11, 14) or ventricular (14) ACh content unless the VNs were stimulated. Stimulation of VNs till exhaustion of the heart response resulted in drop in atrial and ventricular ACh content by ~75% (11, 14). The difference between the isolated and in situ atria could depend on the better availability of choline from the blood in the later.
Taken together, the results of original experiments of the British group (8-10) and of those with HC3 (11) suggested the following picture of the ACh turnover in the working heart. ACh is permanently synthesized in and released from the beating myocytes. The much smaller amount of ACh is synthesized in the parasympathetic fibres and released upon their stimulation (Fig. 1). As suggested by experiments of Bulbring and Burn (8) and experiments with HC3 in the in situ hearts (11, 14), synthesis and release of the non-neuronal fraction of ACh may be intensified by ACh released from the parasympathetic fibres. This would provide the amplification mechanism in the scarcely parasympathetically innervated ventricles. This hypothesis found confirmation and further development in the recent works referred in the following parts of this review.
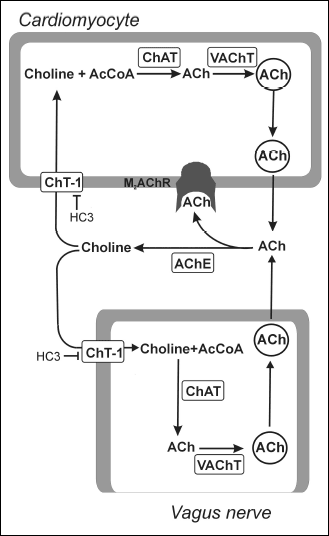 |
Fig. 1. The parallel systems of acetylcholine synthesis, intracellular transport, and release in the postganglionic vagus nerve fibres and in the cardiac myocytes. ACh, acetylcholine; M2AChR, M2 acetylcholine receptor; AcCoA, acetyl-coenzyme A; ChAT, choline acetyltransferase; VAChT, vesicular ACh transporter; AChE, acetylcholine esterase; CHT-1, high-affinity choline transporter (CHT-1); HC3, hemicholinium No 3. |
The advent of modern methods enabled investigations which proved that the heart myocytes synthesize and release ACh. Unfortunately, the recent authors did not mention the above referred earlier investigations maintaining that nothing was known of the non-neuronal ACh in the heart till their own works.
Rana et al. (15) investigated the expression at the mRNA and protein level of all elements of ACh system in the myocytes isolated from different parts of the hearts of the neonatal, young and old rats. The contamination of myocytes by neuronal elements was excluded by the absence of proteins specific for neurofilaments.
The ChAT, VAChT and CHT-1 expression was detected in both atrial and ventricular myocytes (Fig. 1). There were age- and tissue-dependent differences in expression of these proteins. They were not detected in the myocytes of the neonatal hearts. Expression of CHT-1 in atrial myocytes was higher in old then in young rats, while in ventricular myocytes it was lower in the old rats. The regional- and age-dependent VAChT expression was inversely proportional to the CHT-1 expression. ChAT expression was lower in older then in young rats in atrial and ventricular myocytes.
Expression of CHT-1 and VAChT was also revealed in sarcoplasm of left ventricular myocytes of adult rats by immunofluorescence staining. As could be expected, the CHT-1 fluorescence signal was mainly detected close to the sarcolema. Incubation of adult atrial and ventricular myocytes with the blocker of AChE resulted in accumulation of ACh in the supernatant. This shows that myocytes are able to synthesize and secrete ACh.
Expression at mRNA and protein level of ChAT, VAChT and CHT-1 in mice and rat cardiomyocytes was found also by other investigators (16, 17). In Kakinuma et al. (16) experiments ChAT and VAChT proteins were distributed throughout the cytoplasm. Immunogold electron microscopy demonstrated vesicle like structures with VAChT immunoreactivity. In Rocha-Resende et al. (17) experiments ChAT and VAChT were localized mainly in the perinuclear region. VAChT colocalized with the recycling vesicles. These results clearly show that cardiac myocytes possess the set of the proteins necessary for ACh synthesis and packing it into the cytoplasmic vesicles. Indeed, basal level of ACh was detected in rat cultured myocytes (16). Furthermore, it was found that muscarinic agonists, ACh and pilocarpine, stimulate the myocytes ChAT transcriptional activity (16) and thus ACh synthesis (16, 17), these effects being inhibited by atropine, the blocker of M2AChR. Kakinuma et al. (16) proposed that stimulation of expression of ChAT and ACh synthesis by muscarinic agonists may provide an amplification mechanism explaining the effects of VNs in the ventricles sparsely innervated by cholinergic fibres. Moreover, as suggested by our results (11, 14) the release of non-neuronal ACh from the myocytes by ACh released from the parasympathetic fibres may provide the rapidly working amplification mechanism initiating and supporting the mechanism proposed by Kakinuma et al (16).
The recent data beautifully confirm the Bulbring and Burn (8, 9) results and hypothesis that cardiac myocytes are able to synthesize and release ACh and that exogenous ACh may stimulate the myocytes ACh synthesis.
WHAT IS THE ROLE OF NON-NEURONAL ACETYLCHOLINE IN THE HEART?
The most direct way to answer this question is to block chronically the myocytes ACh synthesis and release, and look for the consequences. This was done by Roy et al. (18) in the mice knocked-out of the VAChT gene (VAChT KO mice). Elimination of VAChT expression was cardiomyocyte specific and was intact in the parasympathetic ganglia and fibres. ACh synthesis and secretion was proved in a series of the convincing experiments in myocytes of the wild-type (WT) control mice but was not detectable in the VAChT KO mice.
The in situ hearts of the VAChT KO mice responded to the mild stress (i.p. saline injection) or treadmill exercise test with greater increase in the heart rate and its slower recovery thereafter then the hearts of WT mice. This suggests the imbalance of the autonomic control of the hearts in VAChT KO mice. Under the baseline conditions the hemodynamic parameters of the LV were not changed in VAChT KO mice. However, following the isoproterenol i.v. injection the hearts of VAChT KO mice showed unexpected decrease in the heart rate, in the positive and negative LV dP/dt and decreased heart contractility index.
Moreover, the cardiomyocytes of the VAChT KO mice were hypertrophied. Hypertrophy was also observed in the cultured neonatal myocytes in which VAChT activity was eliminated pharmacologically or by gene silencing with siRNA. The same effect was obtained when ACh synthesis was blocked with HC3. The hypertrophied myocytes displayed increase in the ROS production, and in the peak Ca2+ transients. Expression of the hypertrophy markers, β-myosin heavy chain and atrial natriuretic peptide (ANP) was also increased. These findings are consistent with the earlier results of Rocha-Resende et al. (17) who found that accumulation of ACh in the isolated myocytes by blocking of AChE could offset their hypertrophy stimulated by isoproterenol.
The results of Roy et al. (18) clearly show that cardiomyocyte derived ACh is important for the maintaining of the parasympathetic-sympathetic balance of the autonomic heart control. Moreover, remodeling and hypertrophy of the hearts of VAChT KO mice suggest that non-neuronal ACh is involved in regulation of the important cellular signaling pathways.
ACETYLCHOLINE PROTECTS THE ISCHEMIC/REPERFUSED HEART
The VNs stimulation during temporary ischemia induced in rats and pigs markedly reduced infarct size (~50%), improved LV hemodynamics and reduced incidence of lethal arrhythmias at reperfusion (19-21). These effects were related to attenuation of mitochondrial injury evidenced by swelling, increased reactive oxygen species (ROS) production, depolarization of mitochondrial membranes, and cytochrome c release (21). It has been shown that VNs stimulation in the rat hearts, isolated with their VNs attached, inhibited opening of the mitochondrial permeability transition pore at reperfusion after global ischemia (19). These results rise the question about the mechanisms of these protective effects of VNs stimulation and about the involvement of the non-neuronal ACh production.
The heart possesses a self-defense system against stress of ischemia and hypoxia mediated by hypoxia-inducible factor (HIF-1α). The HIF-1α is a transcription factor activating expression of the genes involved in glycolysis, angiogenesis and adaptation against hypoxic stress (22, 23). The level of HIF-1α may be regulated also independently of hypoxia by ACh via the M2 AChR/PI3K/Act pathway (24). Further, it was found that HIF-1α induces expression of apoptosis inhibitor gene (AI) which is involved in suppressing mitochondrial function, thereby decreasing the oxygen demand of the cells protecting them against hypoxia damage (25).
Kakinuma et al. (26) provided convincing evidence that it is non-neuronal heart’s ACh system that is essential for anti-ischemic protection. They developed the strain of mice overexpressing the heart specific ChAT (ChAT-TG mouse). The TG mice expressed ChAT protein and, importantly, HIF-1α protein to the very high degree and the ACh level in their ventricles was over 50 times higher then in WT mice. The basal hemodynamics did not differ between the ChAT-TG and WT mice, however, the former revealed much higher resistance to anoxia. The size of infarct induced by the coronary artery ligation was largely reduced in ChAT-TG mice. Fourteen days after infarction the heart to body ratio was decreased in ChAT-TG mice as compared with the WT mice suggesting that TG mice were less susceptible to post-infarction remodeling. Survival rate was much higher in TG mice. Also the hearts, isolated from the ChAT-TG mice, showed much higher resistance to global ischemia then the hearts of WT mice. Mitochondrial energy metabolism was suppressed in cultured neonatal myocytes of ChAT-TG mice by AI, the expression of which was induced by high levels of HIF-1α. F1-α stimulated also expression of glucose transporter Glut-4 resulting in increased glucose content in the myocardium of ChAT-TG mice. Thus, the energetic metabolism of the hearts of ChAT-TG mice was switched by increased non-neuronal ACh synthesis from oxidative towards glycolytic one, rendering them more resistant to hypoxia. The hearts of ChAT-TG mice showed also enhanced neoangiogenesis.
The results referred so far suggest the following mechanism of antyischemic protective effects of VNs stimulation. Relatively small amounts of ACh released from the cholinergic VNs endings stimulate synthesis and release of larger amounts of ACh from the myocytes (11, 16). This leads to activation of the HIF-1α /AI pathway as reported by Kakinuma et al. (26).
The VNs stimulation provides also additional protecting mechanism against ischemia/reperfusion injury through anti-inflammatory action. As shown by Zhang et al. (27) VNs stimulation applied in advance and during reperfusion of the in situ dog heart prevented increase in concentration of the pro-inflammatory cytokines such as TNF-α and IL-6 in the blood. The cytokines concentration negatively correlated with ACh level in the serum. VNs stimulation also largely prevented neutrophil infiltration of the ischemic/reperfused LV myocardium.
The VNs stimulation was also shown to largely ameliorate ventricular tachycardia and fibrillation which are the constant feature of reperfusion after myocardial ischemia. These potentially lethal arrhythmias are promoted by impairment of myocardial conduction. The conduction depends on intercellular electrical coupling by connexons, the intercellular channels built of proteins called connexins (Cx). The proper function of the ventricular connexons is secured by phosphorylation of the Cx43 (the protein forming ventricular connexons). In the ischemia and reperfusion Cx43 is dephosphorylated which results in the intercellular uncoupling. The VNs stimulation prevents dephosphorylation of Cx43 (20, 21) securing intercellular conduction and preventing arrhythmias.
ACETYLCHOLINE AND CHRONIC HEART FAILURE, PRE-CLINICAL STUDIES
It has been for long recognized that the imbalance of the autonomic nervous control of the heart is an important feature of the chronic heart failure. It is characterized by excessive activation of the sympathetic and inhibition of the parasympathetic system (28-30). This lead to the idea that restitution of the autonomic balance might be beneficial. The idea has been tested in animal models of the heart failure as well as under the clinical settings.
Chronic stimulation of the right VN in conscious rats after myocardial infarction suppressed mortality rate (14% vs. 50%), markedly inhibited post-infarction hypertrophy and improved LV hemodynamics (31). The level of plasma norepinephrine was by more then 50% lower in rats in which VNs was stimulated as compared with not stimulated controls. The authors proposed that one mechanism of the beneficial effects of the VNs stimulation could be suppression of norepinephrine release from the intraventricular sympathetic fibres. Indeed, it has been proved that VNs stimulation suppresses enhanced norepinephrine release within the area of acute ischemia of the cat left ventricular myocardium (32).
Hamman et al. (33) have shown in dogs with the chronic ischemic heart failure induced by embolization of coronary vessels that VNs stimulation improve significantly LV ejection fraction, and decrease end diastolic and systolic LV volumes, suggesting improvement of LV contractility. Accordingly, plasma and tissue biomarkers of heart failure, pro-atrial natriuretic peptide and C-reactive protein were decreased. The VNs stimulation also significantly ameliorated the inflammatory response evidenced by decrease in the level of TNF-α and IL-6 and increased NO synthesis in LV myocardium. VNs stimulation was below the threshold of bradycardia, so lowering of the heart rate could not be important mechanism of beneficial effects of the therapy. Rather, the increase in NO production, anti-inflammatory effect and possible reduction in ROS production are considered by the authors (33).
Somewhat less encouraging results of chronic VNs stimulation were obtained by Yu et al. (34) in canine model of heart failure caused by the chronic mitral regurgitation. The VNs were stimulated between 3rd and 6th month after surgery. The dogs subjected to the VNs stimulation had higher cardiac output, LV stroke volume, and end-systolic stiffness constant as compared with those with non-stimulated VNs. Their NT-proBNP and C-reactive protein decreased. However, in contrast to the Hamann et al. (33) results, LV ejection fraction, LV end-diastolic dimension, LV stroke volume, and myocyte cross-sectional area did not differ between the groups. The different models of heart failure used in experiments of Haman et al. (33) and of Yu et al. (34) could account for this discrepancy.
The role of non-neuronal ACh in the heart protection was not analyzed in the above referred papers. However, it is conceivable that ACh released from the cholinergic fibres stimulated synthesis and release of ACh in the atrial and ventricular myocytes (16, 17) amplificating the effect of VNs stimulation.
The molecular pathways by which VNs stimulation and/or myocytes endogenous ACh synthesis exert beneficial effects and cells protection are nor yet quite clear. Certainly they all depend on activation of the cardiomyocyte M2AChR since they are inhibited by its blockers. One possible pathway leads to pleiotropic activity of proteinkinase G (PKG). Activation of M2AChR results in increased production and secretion of the atrial natriuretic peptide (ANP) (35) and of the NO (36) in the heart myocytes. NO activates the soluble endoplasmic guanylyl cyclase (sGC) and ANP activates particular guanylylcyclase located in sarcolema (pGC). Both enzymes produce cyclic guanylyl monophosphate (cGMP) which activates PKG (Fig. 2). Among numerous activities PKG exerts an anti-hypertrophic effect and protects myocytes against the hypoxic/ischemic injury (37). Indeed, chronic inhibition of NO synthesis by NG-nitro-l-arginine methyl ester (L-NAME) which would compromise PKG activation, results in cardiac myocytes hypertrophy in mice (38).
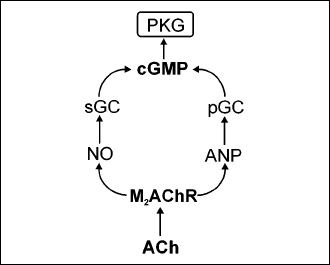 |
Fig. 2. A possible way of the protective effects of acetylcholine in cardiac myocytes by the proteinkinase G activation. ACh, acetylcholine; M2AChR, M2 acetylcholine receptor; NO, nitric oxide; ANP, atrial natriuretic factor; sGC, soluble guanylyl cyclase; pGC, particular guanylyl cyclase; cGMP, cyclic guanylyl monophosphate. |
ACETYLCHOLINE AND THE CHRONIC HEART FAILURE: CLINICAL STUDIES
The results of preclinical studies on the chronic VNs stimulation in the experimental heart failure rised the hope that it might be also beneficial under the clinical settings. This has been initially checked in a non-randomized study of 32 patients with LV systolic heart failure receiving direct VNs chronic stimulation by means of implanted electrode connected to an implanted programmable stimulator. The results seemed promising as VNs stimulation improved the quality of life, exercise capacity, NYHA class, LV ejection fraction and LV systolic volume (39, 40).
Unfortunately, recently published report of the first randomized controlled trial NECTAR-HF (NEural Cardiac TherApy foR Heart Failure) is much less optimistic (41, 42). Eighty seven patients having the implanted VNs stimulation device were randomized at 2:1 ratio to receive stimulation or control (no stimulation) for the 6-months period. After 6 months of VNs stimulation the quality of life was improved as compared with the control group. Moreover, 62% of patients of the VNs stimulation group improved NYHA functional class compared with 45% of control patients. The authors advise to interpret these results with caution because of imperfect patient-level blinding (a fraction of them might guess to which group they were assigned). However, the primary end point, LV end-systolic diameter, and secondary end points: LV end-diastolic diameter, LV end-systolic volume, LV end-diastolic volume, and LV ejection fraction did not differ between the groups. Exercise capacity (peak VO2) and NT-proBNP also did not differ between the groups. Thus, the beneficial effect of VNs stimulation in this group of patients on the progress of systolic heart failure and LV remodeling looks dubious. Discussing the reasons of discrepancies between the results of previous clinical studies and those of NECTAR-HF, the authors mention the inclusion of sham-treated control group in NECTAR-HF and incomplete understanding of appropriate pattern and dosing of VNs stimulation in humans as seemingly most important.
The report concerned the initial 6-months phase, but the investigation is going on. This, and possibly other investigations in large groups of patients should decide whether chronic VNs stimulation in heart failure patients may be beneficial or it will share the fate of numerous methods providing promising results in preclinical studies but failing under the clinical settings.
Conflict of interests: None declared.
REFERENCES
- Wessler I, Kirkpatric CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 2008; 154: 1558-1571.
- Roy A, Guatimosin S, Prado VF, Gros R, Prado MA. Cholinergic activity as a new target in diseases of the heart. Mol Med 2014; 20: 527-537.
- Hoover DB, Ganote CE, Ferguson SM, Blakely RD, Parsons RL. Localization of cholinergic innervation in guinea pig heart by immunochemistry for high-affinity cholinergic transporters. Cardiovasc Res 2004; 62: 112-121.
- Cric SJ, Wharton J, Sheppard MN, et al. Innervation of the human cardiac conduction system. A quantitative immunohistochemical and histochemical study. Circulation 1994; 89: 1697-1708.
- Nachmansohn D, John HM, Berman M. Studies on choline acetylase: the formation of acetylcholine in the nerve axon. J Biol Chem 1946; 163: 475-480.
- Erickson JD, Varogui H, Schafer MK, et al. Functional identification of a vesicular acetylcholine transporter and its expression from a „cholinergic” gene locus. J Biol Chem 1994; 269: 21929-21932.
- Okuda T, Haga T. High affinity choline transporter. Neurochem Res 2003; 28: 483-488.
- Bulbring E, Burn JH. Action of acetylcholine on rabbit auricles in relation to acetylcholine synthesis. J Physiol 1949; 108: 508-524.
- Briscoe S, Burn JH. The formation of an acetylocholine-like substance by the isolated rabbit heart. J Physiol 1954; 126: 181-190.
- Day M. The release of substances like acetylcholine and adrenaline by the isolated rabbit heart. J Physiol 1956; 134: 558-568.
- Lewartowski B, Bielecki K. The influence of hemicholinium No 3 and vagal stimulation on acetylcholine content in rabbit atria. J Pharmacol Exp Ther 1963; 142: 24-30.
- Schueler FW. A new group of respiratory paralysants. I The „hemicholiniums”. J Pharmacol Exp Ther 1955; 115: 127-143.
- Lewartowski B. Selective stimulation cardiac postganglionic fibres. Nature 1963; 199: 76-77.
- Bielecki K, Lewartowski B. The influence of hemicholinium No 3 and vagus stimulation on acetylcholine distribution in the cat’s heart. Pflugers Archiv 1964; 279: 149-55.
- Rana OR, Schauerte P, Kluttig R, et.al. Acetylcholine as an age-dependent non-neuronal source in the heart. Auton Neurosci 2010; 156: 82-89.
- Kakinuma Y, Akiyama T, Sato T. Cholinoceptive and cholinergic properties of cardiomyocytes involving amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J 2009; 276: 5111-5125.
- Rocha-Resende C, Roy A, Resende R, et al. Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J Mol Cell Cardiol 2012; 53: 206-216.
- Roy A, Fields WC, Rocha-Resende C, et al. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J 2013; 27: 5072-5082.
- Katare RG, Ando M, Kakinuma Y, et al. Vagal nerve stimulation prevents reperfusion injury through inhibition of opening of mitochondrial permeability transition pore independent of the bradycardiac effect. J Thorac Cardiovasc Surg 2009; 137: 223-231.
- Ando M, Katare RG, Kakinuma Y, et al. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving connexin 43 protein. Circulation 2005; 112: 164-170.
- Shinlapawittayatorn K, Chinda K, Palee S, et al. Vagus nerve stimulation initiated late during ischemia but not reperfusion exerts cardioprotection via amelioration of cardiac mitochondrial function. Heart Rhythm 2014; 11: 2278-2287.
- Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol 2011; 76: 347-353.
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148: 399-408.
- Kakinuma Y, Ando M, Kuwabara M, et al. Acetylcholine from vagal simulation protects cardiomyocytes against ischemia and hypoxia involving additive non-hypoxic induction of HIF-1alpha. FEBS Lett 2005; 579: 2111-2118.
- Kakinuma Y, Katare R, Arikawa M, et al. A HIF-1alpha-related gene involved in cell protection from hypoxia by suppression of mitochondrial function. FEBS Lett 2008; 582: 332-340.
- Kakinuma Y, Tsuda M, Okazaki K, et al. Heart-specific overexpression of choline acetyltransferaze gene protects murine heart against ischemia through hypoxia-inducible factor-1alpha related defense mechanism. J Am Heart Assoc 2013; 2: e004887.
- Zhang R, Wugeti N, Sun J, et al. Effects of vagus nerve stimulation via cholinergic anti-inflammatory pathway activation on myocardial ischemia/repefusion injury in canine. Int J Clin Exp Med 2014; 7: 2615-2623.
- Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ. Parasympathetic withdrowal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol 1991; 18: 464-472.
- Schwartz PJ, De Ferrari GM. Sympathetic-parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail Rev 2011; 16: 101-107.
- Florea VG, Cohn JN. The autonomic nervous system and heart failure. Circ Res 2014; 114: 1815-1826.
- Li M, Zhen MS, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation 2004; 109: 120-124.
- Kawada T, Yamazaki T, Akiyama T, et al. Vagal stimulation suppresses ischemia-induced myocardial interstitial norepinephrine release. Life Sci 2006; 78: 882-887.
- Hamann JJ, Ruble SB, Stolen C, et al. Vagus nerve stimulation improves left ventricular function in a canine model of chronic heart failure. Eur J Heart Fail 2013; 15: 1319-1326.
- Yu H, Tang M, Yu J, Zhou X, Zeng L, Zhang S. Chronic vagus nerve stimulation improves left ventricular function in canine model of chronic mitral regurgitation. J Transl Med 2014; 12: 302-309.
- Kim HY, Cho KW, Xu DY, Kang DG, Lee HS. Endogenous ACh tonically stimulates ANP secretion in rat atria. Am J Physiol Heart Circ Physiol 2013; 305: H1050-H1056.
- Brack KE, Patel VH, Mantravardi R, Coote JH, Ng GA. Direct evidence of nitric oxide release from neuronal nitric synthase activation of the left ventricle as a result of cervical vagus nerve stimulation. J Physiol 2009; 587: 3045-3054.
- Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J 2012; 76: 1819-1825.
- Oscan RJ, Lai YN, Prabhu KV, Hambly BD, McLachlan CS. Chronic NG-nitro-l-arginine methyl estrer (L-NAME) administration in C57BL/6J mice induces a sustained derease in c-kit positive cells during development of cardiac hypertrophy. J Physiol Pharmacol 2013; 64: 727-736.
- Schwartz PJ, De Ferrari GM, Sanzo A, et al. Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Fail 2008; 10: 884-891.
- De Ferrari GM, Crijns HJ, Borggrefe M, et al. Chronic vagus nerve stimulation a new and promising therapeutic approach for chronic heart failure. Eur Heart J 2011; 32: 847-855.
- Zannad F, De Ferrari G, Tuinenburg AE, et al. Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the NEural Cardiac TherApy foR Heart Failure (NECTAR-HF) randomized controlled trial. Eur Heart J 2015; 36: 425-433.
- Camm AJ, Savelieva I. Vagal nerve stimulation in heart failure. Eur Heart J 2015; 36: 404-406.
A c c e p t e d : July 15, 2015