THE INFLUENCE OF PRETREATMENT WITH GHRELIN ON THE DEVELOPMENT OF ACETIC-ACID-INDUCED COLITIS IN RATS
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic relapsing inflammation of the digestive tract with alternate periods of exacerbation and remission. The main forms of IBD are Crohn’s disease and ulcerative colitis (1). Epidemiological studies indicate that the frequency of IBD incidence is rising globally (1-5). The etiology of IBD is still unclear, but research in basic science, experimental animal models, genetics and human clinical trials has provided new data concerning the pathogenesis of bowel inflammation. It is currently suggested that IBD results from an abnormal immunological response to micro flora present in the digestive system and their pathogenesis is complex, and requires co-existence of factors of environmental and genetic nature (1, 5, 6). There are numerous methods to treat IBD, but medication that would ensure permanent therapeutic effect has not been found so far (1, 6, 7). Therefore, it is vital to search for the new therapeutic strategies.
Ghrelin, a 28-amino acid peptide, discovered in human and rat stomachs (8-10), is an endogenous ligand for a receptor activated by factors which stimulate growth hormone secretion (growth hormone secretagogue receptor (GHS-R)). The receptor is present mainly in the pituitary gland and hypothalamus, but also in other tissues of the body (11). Besides inducing body mass growth through food intake stimulation and a decrease in utilization of fat tissue (12), ghrelin demonstrates anti-inflammatory properties. Receptors for ghrelin are present, among others, on the immunological system cells, suggesting that ghrelin may modulate inflammatory response (13).
Previous studies have shown ghrelin exhibits protective and therapeutic on the gut. Pretreatment with ghrelin inhibits the development of experimental gastric ulcers induced by ethanol (14), stress (15) or alendronate (16). Moreover, administration of ghrelin accelerates the healing of gastric (17, 18), duodenal (17, 19) and oral (20) ulcers evoked by different noxious agents. Protective and therapeutic effect of ghrelin has been also found in the pancreas. Pretreatment with ghrelin inhibits the development of cerulein- and ischemia/reperfusion-induced pancreatitis (21, 22) and accelerates its healing (23-25).
The role of ghrelin in IBD is not clear. Clinical studies have shown that expression of mRNA of ghrelin in colonic mucosa is significantly upregulated in patients with IBD and correlated with the grade of inflammation (26, 27). Moreover, patients with active phase of IBD have higher serum ghrelin concentration than patients in remission or healthy individuals and the level of circulating ghrelin correlates with the severity of IBD (28-30).
There are animal experimental studies concerning the role of ghrelin in the development and healing of colitis. Some studies have shown that administration of exogenous ghrelin significantly accelerates the healing of colitis evoked by trinitrobenzene sulfonic acid (TNBS) in mice (31) and rats (26). Therapeutic effect of ghrelin administration has also been found in dextran sulfate sodium (DSS)-induced colitis in rats (32). In contrast to those data are results obtained by De Smet et al. (33). They have reported that endogenous and exogenous ghrelin enhances the colonic manifestation of DSS-induced colitis in mice. Pro-inflammatory properties of ghrelin in colitis have been also postulated by Zhao et al. (34). They have found that ghrelin stimulates interleukin-8 gene expression through protein kinase C-mediated NF-kappaB pathway in human colonic epithelial cells.
The objective of present study was determine the influence of pretreatment with ghrelin on the development of acetic acid-induced colitis in rats. The possible protective effect of ghrelin in the large intestine could be useful in clinical condition in maintaining the IBD patients in remission.
MATERIALS AND METHODS
Animals and treatment
The research was performed on Wistar male rats weighing 250 – 270 g and conducted following the experimental protocol approved by the First Local Commission of Ethics for the Care and Use of Laboratory Animals in Cracow. During study, the animals were kept in cages placed in temperature and 12-hour light-darkness cycle was maintained. The animals, prior to colitis induction with acetic acid solution, were starved for 18 hours with free access to water. Earlier and after this period food and tap water were available ad libitum.
One hundred sixty rats were randomly divided in eight equal experimental groups: [1] control rats treated intraperitoneally (i.p.) with saline without induction of colitis; [2-4] rats treated i.p. with ghrelin given at the dose of 4, 8 or 8 nmol/kg/dose without induction of colitis; [5] rats treated i.p. with saline before induction of colitis; [6-8] rats treated i.p. with ghrelin given at the dose of 4, 8 or 16 nmol/kg/dose before induction of colitis.
Animals from each experimental group were divided into two equal sub-groups. In the first sub-groups, the severity of colitis was assessed after 1 h from acetic acid enema. In the second sub-groups, the severity of colitis was assessed after 24 h from acetic acid enema.
Experiments were repeated to obtain 10 animals in each experimental group and each time of observation.
According to group of animals, saline or rat ghrelin (Yanaihara Institute, Shizuoka, Japan) were administered intraperitoneally twice, 8 and 1 h before rectal administration of saline or acetic acid solution. Ghrelin was dissolved in saline and then administered in an amount which did not exceed 0.3 ml/dose.
Before induction of colitis, animals were anesthetized with ketamine (50 mg/kg i.p., Bioketan, Vetoquinol Biowet, Gorzow Wielkopolski, Poland). Colitis was induced by intrarectal administration of 1 ml of 4% acetic acid aqueous solution through a polyethylene catheter, which the end was inserted into the bowel by 4.5 cm from the anus. Animals without induction of colitis were treated with rectal enema of saline (sham-operation).
Measurement of colonic blood flood and mucosal lesions
One or 24 h after rectal enema, rats were anesthetized again with ketamine. After opening the abdominal cavity and exposing the colon, the measurement of colonic blood flow volume was performed using laser Doppler flowmeter (PeriFlux 4001 Master monitor, Perimed AB, Jarfalla, Sweden), in accordance with the methodology described before (35). The blood flow measurement was performed every time in five various parts of the descending and sigmoid colon and a mean value of five records was expressed as the percentage of value obtained in animals from the control group. After measurement of colonic blood flow, the area of mucosal damage was measured, using a computerized planimeter (Morphomat, Carl Zeiss, Berlin, Germany), in accordance to the method described earlier (19).
Biochemical analysis
After measurement of colonic blood flow and lesions area a fragment of colonic wall for histological examination was cut out by scissors. After that mucosa from remaining parts of the colon was scrapped using a glass microscope slides over a glass plate. Then samples of mucosa from the colon were divided for determination of mucosal DNA synthesis (an index of mucosal cell proliferation), concentration of pro-inflammatory interleukin-1β, activity of myeloperoxidase, concentration of malondialdehyde (MDA) (as an index of lipid peroxidation) and activity of superoxide dismutase (SOD).
Determination of DNA synthesis in colonic mucosa
DNA synthesis was determined by measurement of [3H]thymidine incorporation ([6-3H]-thymidine, 20 – 30 Ci/mmol, Institute for Research, Production and Application of Radioisotopes, Prague, Czech Republic) into mucosal DNA as described previously (36). The incorporation of labeled thymidine into DNA was determined by counting 0.5 ml DNA-containing supernatant in a liquid scintillation system. The rate of DNA synthesis was expressed as tritium disintegrations per minute per µg of DNA (dpm/µg DNA).
Determination of interleukin-1β concentration in colonic mucosa
Samples of colonic mucosa were homogenized in ice-cold phosphate buffered saline (PBS, 20 mM, pH 7.4). Homogenate was centrifuged at 1500 g for 10 min at 4°C. Content of interleukin-1β in the supernatant was measured using the BioSource Cytoscreen rat IL-1β kit (BioSource International, Camarillo, California, USA) based on ELISA. Concentration of interleukin-1β in colonic mucosa was expressed as ng per g of tissue.
Determination of myeloperoxidase activity in colonic mucosa
Samples of colonic mucosa were frozen in liquid nitrogen, and, until the marking was done, stored at the temperature of –60°C. Myeloperoxidase activity was assessed using a modification of the method described by Bradley et al. (37). Mucosa was homogenized in 1 ml of 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% of hexadecyltrimethyl ammonium bromide. Then the homogenate was freeze-thawed three times, subjected to sonication in ice bath for 20 s and centrifuged at 3000 g for 15 min at the temperature of 4°C. 100 µl of the supernatant was taken and 2.9 ml of 50 mM phosphate buffer was added, which contained 157 µg/ml of o-dianisidine dihydrochloride and 0.0005% of hydrogen peroxide. Hydrogen peroxide reduction through myeloperoxidase causes the oxidation of o-dianisidine and produces a stained final product. The intensity of staining was measured spectrophotometrically with a light wave of the length of 460 nm. The obtained results were calculated in units per gram of tissue and finally expressed as the percentage of the value observed in the control group.
Determination of malondialdehyde concentration in colonic mucosa
Peroxidation of lipids in colonic mucosa was tested by measurement of MDA concentration using commercial kit Bioxytech ®LPO-586TM (OxisResearchTM, OXIS Health Products, Inc., Portland, OR, USA), as described previously (38). Prior to homogenization of tissue samples, 10 µl 0.5 M butylated hydroxytoluene in acetonitrile was added to prevent sample oxidation during homogenization. Mucosa was homogenized in ice-cold Tris buffer (20 mM, pH 7.4), centrifuged (3000 g at 4°C for 10 min) and supernatant was used for the assay. Results were expressed in nanomoles per gram of colonic mucosa.
Determination of superoxide dismutase activity in colonic mucosa
To determine the activity of SOD in colonic mucosa, tissue was homogenized in 20 mM HEPES buffer, pH 7.2, containing 1 mM EGTA, 210 mM mannitol, and sucrose. Homogenate was centrifuged at 1500 g for 5 min at 4°C. Activity of SOD in the supernatant was measured using Superoxide Dismutase Assay Kit (Cayman Chemical Company, Ann Arbor, MI, USA). Results have been expressed in units per g of colonic mucosa.
Histological examination of the colon
Samples of the colon were fixed in 10% buffered formaldehyde and embedded in paraffin. Paraffin sections were stained with hematoxylin and eosin. Slides were examined by two experienced pathologists without knowledge of the treatment given. The histological grading of colonic damage was determined using a scale previously presented by Vilaseca (39). The histological grading of lesions was made using scale ranging from 0 to 2 (0 = no lesions; 1 = small lesions < 3mm; 2 = large lesions > 3 mm). Inflammatory infiltration was graded from 0 to 3 (0 = none; 1 small; 2 = moderate; 3 = heavy), depth of the lesions was graded from 0 to 3 (0 = no lesions; 1 = lesions reaching submucosa; 2 = lesions reaching muscularis propria; 3 = lesions reaching serosa). The presence of fibrosis was grading from 0 to 2 (0 = none; 1 = mild; 2 = severe). Moreover, the presence of arterial vessels inflammation was assessed.
Statistical analysis
Results were presented as a mean value ± standard error (S.E.M.). Statistical assessment was done through one way analysis of variance followed by Tukey’s multiple comparison test using GraphPadPrism (GraphPad Software, San Diego, CA, USA). Differences were considered to be statistically significant if P was less than 0.05.
RESULTS
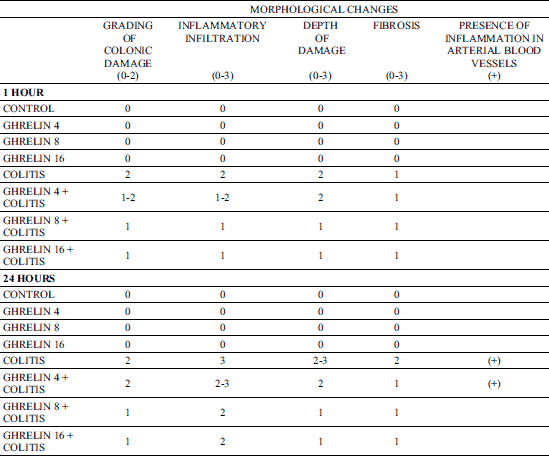
Macroscopic and microscopic morphological features obtained from rats treated with saline (control) or ghrelin without induction of colitis, did not show any damage of the colon (Fig. 1, Table 1). That lack of damage was found in both times of observation, 1 and 24 hours after enema with saline. Rectal administration of 4% solution of acetic acid caused the development of colitis in all rats. One hour after administration of acetic acid, the average area of colonic lesions was 5.0 ± 0.3 mm2; whereas 23 h later the area of colonic lesions enlarged to 28.6 ± 1.4 mm2 (Fig. 1). In animals with colitis, histological examination performed 1 hour after acetic acid enema, showed the presence of large lesions reaching the level of muscular membrane, with moderate inflammatory cell infiltration (Table 1). Twenty three hour later, histological examination showed an increase in colonic damage. There were large ulcers, reaching the muscular membrane or, in half of cases, even serous membrane of the colonic wall. These changes were accompanied by heavy inflammatory leukocytic infiltration and severe fibrosis together with arterial vessels inflammation (Table 1, Fig. 2). Pretreatment with ghrelin reduced the development of colitis. One hour after acetic acid enema, the area of colonic damage was reduced by 10, 56 or 60% in rats pretreated ghrelin given at the dose of 4, 8 or 16 nmol/kg/dose, respectively. Twenty three hours later in rats pretreated with ghrelin given at the dose of 4, 8 or 16 nmol/kg/dose, the area of colonic lesion was reduced by 8, 50 or 56% (Fig. 1). The effect caused by ghrelin given at the dose of 8 and 16 nmol/kg/dose was statistically significant in both times of observation, 1 and 24 hours after enema with acetic acid solution.
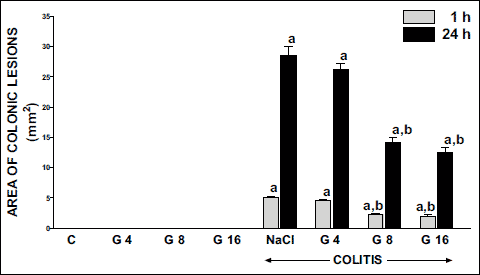 |
Fig. 1. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on the area of colonic lesions observed 1 or 24 h after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
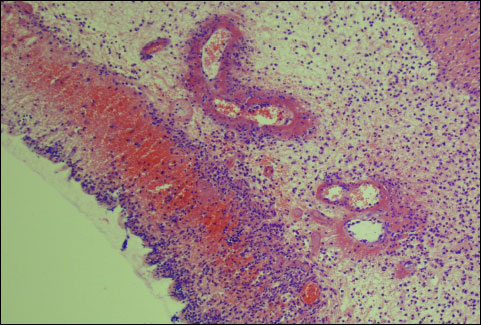 |
Fig. 2. Morphological signs of colonic damage observed 24 hours after rectal enema with acetic acid. Hematoxylin-eosin stain, original magnification ×200. |
Pretreatment with ghrelin reduced also histological manifestation of colonic damage evoked by acetic acid (Table 1, Fig. 3). One hour after enema with acetic acid, beneficial effect of pretreatment with ghrelin was found as a decrease in grading and depth of ulcers, and reduction in inflammatory infiltration of the colonic wall. Twenty three hours later, the beneficial effect of pretreatment with ghrelin was manifested, apart effects mentioned above, by a reduction in fibrosis and prevention of inflammation in arterial blood vessels (Table 1).
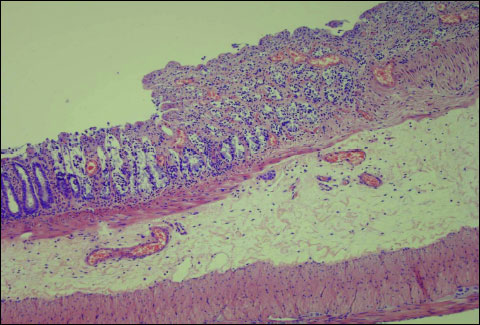 |
Fig. 3. Influence of pretreatment with ghrelin given at the dose of 8 nmol/kg/dose on morphological signs of colonic damage observed 24 hours after rectal enema with acetic acid. Hematoxylin-eosin stain, original magnification ×200. |
In the animals from the control group, the rate of DNA synthesis in colonic mucosa reached a value of 35.1 ± 1.4 or 36.2 ± 1.3 dpm/µg DNA at the time of 1 or 24 h after saline enema, respectively (Fig. 4). Pretreatment with ghrelin at the dosage used, tended to increase DNA synthesis in colonic mucosa in rats without induction of colitis (Fig. 4), but this effect was statistically insignificant (Fig. 4). Induction of colitis through rectal administration of 4% solution of acetic acid inhibited DNA synthesis in the colon. One and 24 h after administration of acetic acid, DNA synthesis was significantly reduced by around 40 and 60%, respectively. Pretreatment with ghrelin before induction of colitis partly reversed that effect. In both times of observation, ghrelin given at the dose of 4 nmol/kg/dose tended to increase mucosal DNA synthesis, but that effect was statistically insignificant (Fig. 4). In contrast to that, ghrelin administered at the dose of 8 or 16 nmol/kg/dose, led to a statistically significant and similar in case of both doses, improvement of DNA synthesis in the colon mucosa in animals with induction of colitis. This effect was observed after 1 and 24 hours after induction of colitis (Fig. 4).
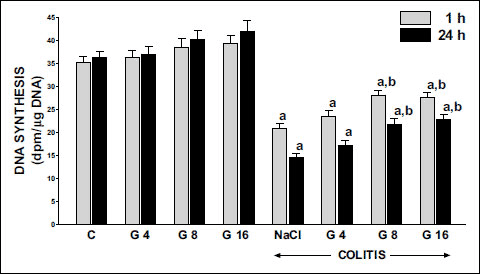 |
Fig. 4. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on DNA synthesis in colonic mucosa observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
Double intraperitoneal administration of ghrelin, in doses used, had no influence on mucosal blood flow in the colon in animals without induction of colitis (Fig. 5). Induction of colitis led to a statistically significant decrease in colonic mucosal blood flow. One hour after induction of colitis mucosal blood flow in the colon was reduced by almost 68%, when compared to a value observed in the animals from the control group (Fig. 5). Twenty three hours later, mucosal blood flow in the colon was reduced by nearly 75%. Pretreatment with ghrelin given at the dose of 8 or 16 nmol/kg/dose partly, but significantly, reversed the colitis-evoked decrease in mucosal blood flow in the colon. This effect of ghrelin was found in both times of observation, 1 and 24 h after induction of colitis. Effect of ghrelin given at the dose of 4 nmol/kg/dose was weak and statistically insignificant (Fig. 5).
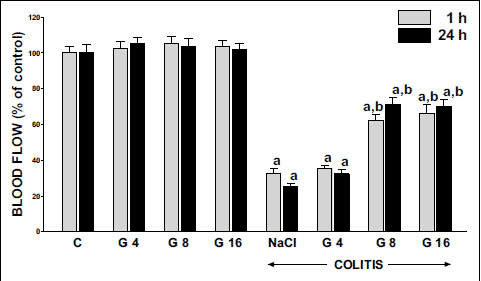 |
Fig. 5. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on mucosal blood flow in the colon observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
The concentration of pro-inflammatory interleukin 1β (IL-1β) in colonic mucosa in control animals was around 0.50 ng/g of tissue (Fig. 6). Double administration of ghrelin, at the dosage used, had no influence on IL-1β concentration in colonic mucosa in animals without induction of colitis. Induction of colitis through rectal administration of 4% acetic acid led to a statistically significant increase in the concentration of IL-1β in colonic mucosa. One hour after induction of colitis, mucosal concentration of IL-1β was increased by more than 100%; whereas 23 hours later almost a 12-fold increase in that pro-inflammatory cytokine was observed (Fig. 6). Administration of ghrelin inhibited the colitis-induced increase in IL-1β concentration in colon mucosa. The influence of ghrelin, given at the dose 8 or 16 nmol/kg/dose, on this parameter in rats with colitis was statistically significant and similar for both of those doses. This effect was observed in both periods of observation, 1 h and 24 hours after induction of colitis. On the other hand, pretreatment with ghrelin administered at the dose of 4 nmol/kg/dose failed to affect the mucosal concentration of IL-1β in the colon of rats with colitis (Fig. 6).
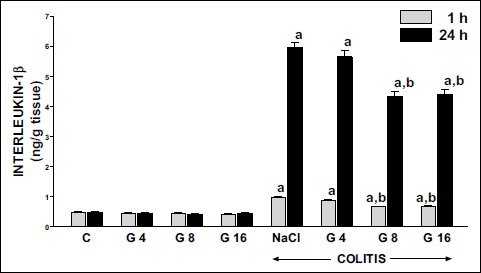 |
Fig. 6. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on interleukin-1β concentration in colonic mucosa observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
In control saline-treated animals, myeloperoxidase (MPO) activity in colonic mucosa was low and reached a value of 0.70 U/g of tissue (Fig. 7). Pretreatment with ghrelin, at the doses used, failed to affect MPO activity in colonic mucosa in rats without induction of colitis. Induction of colitis resulted in a significant increase in MPO activity in colonic mucosa. One h after induction of colitis, MPO activity reached 2-fold increase; whereas 23 hours later more than a 4-fold increase was observed. Pretreatment with ghrelin reduced the colitis-evoked increase in colonic activity of MPO. The effect of ghrelin, administered at the dose of 4 nmol/kg/dose, was statistically insignificant; whereas the two remaining doses inhibited significantly the mucosal MPO activity in the colon of animals with colitis. This inhibitory effect was similar for both doses of ghrelin and was observed ether 1 or 12 hours after induction of colitis (Fig. 7).
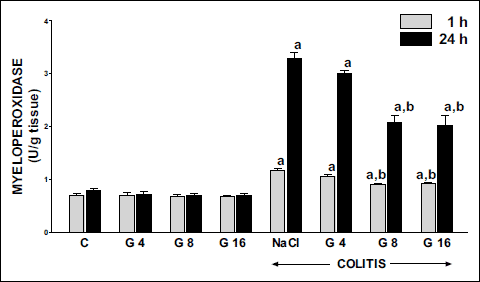 |
Fig. 7. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on myeloperoxidase activity in colonic mucosa observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
A value of malondialdehyde (MDA) concentration in colonic mucosa in control saline-treated rats was between 4.26 and 4.50 nmol/g of tissue (Fig. 8). Pretreatment with ghrelin had no significant influence on MDA concentration in colonic mucosa in animals without induction of colitis. Induction of colitis by rectal administration of 4% solution of acetic acid caused a statistically significant increase in MDA concentration in mucosa of the colon. More than a 3-fold and 5-fold rise in this parameter was observed 1 and 24 h after induction of colitis; respectively. Pretreatment with ghrelin inhibited the colitis-induced increase in MDA concentration in colonic mucosa. In both periods of observation, that effect was statistically significant and reached a similar intensity after ghrelin administered at the dose of 8 or 16 nmol/kg/dose. Ghrelin given at the dose of 4 nmol/kg/dose failed to significantly affect MDA concentration in colonic mucosa in rats with colitis (Fig. 8).
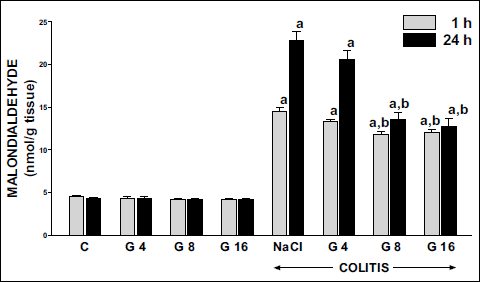 |
Fig. 8. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on concentration of malondialdehyde in colonic mucosa observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
Superoxide dismutase (SOD) activity in mucosa in control saline-treated rats was around 360 U/g of tissue (Fig. 9). Administration of ghrelin in doses used failed to affect mucosal SOD activity in the colon of animals without induction of colitis. Induction of colitis significantly reduced SOD activity and this effect was found in both periods of observation, 1 and 24 hours after induction of colitis, mucosal SOD activity was decreased by 34 and 46%, respectively. Pretreatment with ghrelin partly reversed the colitis-induced reduction in mucosal SOD activity. This effect was statistically significant after ghrelin administered at the dose of 8 or 16 nmol/kg/dose. Ghrelin given at the dose of 4 nmol/kg/dose failed to significantly affect SOD activity in colonic mucosa in rats with colitis (Fig. 9).
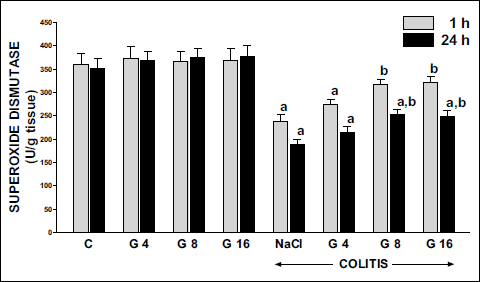 |
Fig. 9. Influence of pretreatment with ghrelin given intraperitoneally at the dose of 4, 8 or 16 nmol/kg/dose (G4, G8 or G16) and colitis evoked by rectal enema with acetic acid on superoxide dismutase activity in colonic mucosa observed 1 or 24 hours after induction of colitis. Mean ± standard error. N = 10 animals in each group. aP<0.05 compared to control (C) at the same time of observation; bP<0.05 compared to NaCl + colitis at the same time of observation. |
DISCUSSION
Our present study has provided several important observations regarding the influence of exogenous ghrelin on the integrity of the colonic mucosa. We have found that pretreatment with ghrelin exhibits protective effect in the colon and inhibits the development of colitis evoked by acetic acid enema. Protective effect of ghrelin was manifested as an improvement of colonic morphology and mucosal blood flow, a decrease in biochemical indexes of inflammation as well as a partial reversion of the colitis-evoked reduction in mucosal cell vitality.
Our study has demonstrated that the area of colonic damage increases more than 5-fold between the 1st and 24th h after acetic acid enema. Pretreatment with ghrelin reduced colonic damage and this effect was observed in both periods of observation. The decrease in colonic damage was manifested as a reduction of the area of damage and an improvement in the colon morphology observed in the microscopic examination. A statistically significant protective effect of ghrelin was observed if this peptide was administered at the dose of 8 or 16 nmol/kg/dose and a rate of this effect was similar for of both above doses. This observation is in agreement with previous studies showing that those doses of ghrelin, 8 or 16 nmol/kg/dose, exhibit protective and/or therapeutic effect in other organs, such as the pancreas (22, 23), stomach (17), duodenum (17) and oral cavity (20).
Cell proliferation in mucosa of the gut exhibits high dynamics and colonic mucosa is renewed every 3 – 8 days (40). Insufficient cell proliferation and/or increased cell loss may lead to mucosal atrophy and the development of ulceration (41, 42). On the other hand, increased cell proliferation may result in hyperplasia, but it is also associated with increased protection of mucosa against damage evoked by noxious agents, as well as accelerates mucosal regeneration after damage of the stomach and intestine (43-46). In synthesis (S) phase of cell-division cycle, DNA is replicated and this process is essential for preparing of the cell to division (47). Our present study has shown that induction of colitis is associated with a decrease in mucosal DNA synthesis in the colon. The rate of mucosal DNA synthesis was inversely proportional to the grade of colonic damage, higher 1 hour after induction of colitis and lower after next 23 hours. In rats without induction of colitis, administration of ghrelin was without a significant influence on DNA synthesis in colonic mucosa. On the other hand, pretreatment with ghrelin, prior to colitis induction with acetic acid, caused a considerable reversal of the colitis-evoked decrease in DNA synthesis in the colon. These findings indicate that protective effect of ghrelin in the colon is related, at least in part, to an increase in vitality of cells in colonic mucosa.
Previous studies have shown that exposure of gastric mucosa to potentially damaging factors results in little or no damage, as long as an adequate blood flow is sustained. On the other hand, a decrease in mucosal blood flow increases the severity and extension of gastric lesions (48). The importance of appropriate blood flow in maintaining mucosal integrity and healing of mucosal damage has been also found in other parts of the digestive tract system such as the oral cavity (20), esophagus (49), duodenum (50) and colon (51). Findings of our present study are in harmony with those data. We have found that induction of colitis reduced mucosal blood flow in the colon and the grade of this reduction was well-correlated with the severity of colitis. Pretreatment with ghrelin prior to colitis induction with acetic acid led to improvement of blood flood through colonic mucosa and that effect was associated with a proportional reduction in the area and severity of colonic damage. This observation indicates that ghrelin’s protective effect on colonic mucosa is connected with an improvement of mucosal blood flow. A question remains, however, whether the ghrelin-evoked improvement of blood flood through colonic mucosa in rats with colitis is a mechanism or a result of ghrelin’s protective effect in the colon. Previous studies have documented that the relation between mucosal blood flow and mucosal damage is bidirectional. Improvement of mucosal blood flow decreases mucosal injury, but simultaneously decreasing the severity of mucosal damage leads to an improvement in the blood flow through mucosal membrane (36, 48, 52, 53).
Another interesting discovery of our present study was the observation regarding the influence of intraperitoneal administration of ghrelin and rectal administration of acetic acid solution on mucosal concentration of interleukin-1b (IL-1β) and myeloperoxidase activity in the colon. IL-1β plays a crucial role in in the induction of local inflammation and systemic acute phase response and in the release of subsequent pro-inflammatory cytokines in the biochemical cascade of inflammation (54, 55). Numerous experimental and clinical studies confirm that administration of IL-1β receptor antagonists or antibodies against IL-1β prevents the increase in serum concentration of pro-inflammatory interleukin-6 and tumor necrosis factor-α, as well as decreases the severity of systemic inflammation (56-60). Our present study has shown that induction of colitis by acetic acid enema leads to a damage and inflammatory infiltration of colonic mucosa, and increases mucosal concentration of IL-1β. On the other hand, pretreatment with ghrelin has reduced mucosal concentration of IL-1β in the colon of rats with colitis. These data indicate that ghrelin reduces a local inflammatory response in acetic acid-induced colitis and this observation seems to show the next mechanism involved in the ghrelin-evoked protective effect in the colon.
Myeloperoxidase (MPO) is released from the granules of neutrophils in inflammatory reactions and for this reason activity of this enzyme reflects the degree of tissue infiltration by neutrophils (61-63). In our present study, induction of colitis increased MPO activity, whereas pretreatment with ghrelin partly, but significantly, reversed this effect. This finding is the next evidence that pretreatment with ghrelin exhibits anti-inflammatory effect in the colon.
Oxidative stress was originally defined as the imbalance between pro-oxidants and antioxidants in biological systems and is a result of increased production of reactive oxygen species (ROS) and/or impaired antioxidant capacity (64). ROS consist of a group of highly reactive oxygen metabolites, including superoxide anion radicals, hydrogen peroxide, hypochlorous acid and the especially reactive hydroxyl radical (65). In physiological condition, the use of oxygen during normal metabolism produces low levels of ROS and excess of ROS are safely neutralized by antioxidant mechanisms involving antioxidant enzymes and scavengers. Superoxide dismutase (SOD), catalase and glutathione peroxidase are the main enzymes responsible for ROS neutralization (66). Oxidative stress plays an important role in the pathogenesis of inflammation. Involvement of ROS has been shown in the pathogenesis of the aspirin- (67), stress- (68) or ischemia/reperfusion-induced gastric lesions (66). Also in the case of inflammatory bowel disease, the role of oxidative stress is being emphasized (69).
In pathology, when ROS production exceeds the capacity of the antioxidant defense system, ROS causes oxidative damage of all cellular elements (64, 70). Peroxidation of cellular lipids, called lipid peroxidation, is the first stage of ROS-mediated cellular damage (65). Our present study has shown that the development of acetic acid-induced colitis is associated with oxidative stress. We have found an increase in concentration of lipid peroxidation product, MDA in colonic mucosa and this effect has been associated with a reduction in SOD activity in this mucosa, leading to additional disruption of redox balance. Pretreatment with ghrelin, prior to rectal administration of acetic acid resulted in a reduction of redox imbalance. Mucosal level of MDA was reduced, whereas mucosal activity of SOD was partly restored. These observations indicate that pretreatment with ghrelin reduces the colitis-induced oxidative stress in colonic mucosa and improves anti-oxidant defense in this inflammation.
Finally, we conclude that pretreatment with ghrelin inhibits the development of the acetic acid-induced colitis and this effect seems to be related to the ghrelin evoked anti-inflammatory and anti-oxidative effects.
Conflict of interests: None declared.
REFERENCES
- Stenson WF, Hanauer SB, Cohen RD. Inflammatory bowel disease. In: Textbook of Gastroenterology, Yamada T, Alpers DH, Kalloo AN, Kaplowitz N, Owyang C, Powell DW (eds). Chichester, Wiley-Blackwell, 2009, pp. 1386-1472.
- Tsironi E, Feakins RM, Probert CS, Rampton DS, Phil D. Incidence of inflammatory bowel disease is rising and abdominal tuberculosis is falling in Bangladeshis in East London, United Kingdom. Am J Gastroenterol 2004; 99: 1749-1755.
- Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142: 46-54.
- Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013; 62: 630-649.
- Ng SC, Tang W, Leong RW, et al. Environmental risk factors in inflammatory bowel disease: a population-based case-control study in Asia-Pacific. Gut 2015; 64: 1063-1071.
- Baumgart DC. The diagnosis and treatment of Crohn’s disease and ulcerative colitis. Dtsch Arztebl Int 2009; 106: 123-133.
- Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet 2012; 380 (9853) :1590-1605.
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone releasing acylated peptide from stomach. Nature 1999; 402: 656-660.
- Ariyasu H, Takaya K, Tagami T, et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab 2001; 86: 4753-4758.
- Ceranowicz P, Warzecha Z, Dembinski A. Peptidyl hormones of endocrine cells origin in the gut - their discovery and physiological relevance. J Physiol Pharmacol 2015; 66: 11-27.
- Gnanapavan S, Kola B, Bustin SA, et al. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab 2002; 87: 2988-2991.
- Tschop M, Flora DB, Mayer JP, Heiman ML. Hypophysectomy prevents ghrelin-induced adiposity and increases gastric ghrelin secretion in rats. Obes Res 2002; 10: 991-999.
- Dixit VD, Schaffer EM, Pyle RS, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest 2004; 114: 57-66.
- Sibilia V, Rindi G, Pagani F, et al. Ghrelin protects against ethanol-induced gastric ulcers in rats: studies on the mechanisms of action. Endocrinology 2003; 144: 353-359.
- Brzozowski T, Konturek PC, Konturek SJ, et al. Exogenous and endogenous ghrelin in gastroprotection against stress-induced gastric damage. Regul Pept 2004; 120: 39-51.
- Iseri SO, Sener G, Yuksel M, et al. Ghrelin against alendronate-induced gastric damage in rats. J Endocrinol 2005; 187: 399-406.
- Ceranowicz P, Warzecha Z, Dembinski A, et al. Treatment with ghrelin accelerates the healing of acetic acid-induced gastric and duodenal ulcers in rats. J Physiol Pharmacol 2009; 60: 87-98.
- Warzecha Z, Ceranowicz P, Dembinski M, et al. Involvement of cyclooxygenase-1 and cyclooxygenase-2 activity in the therapeutic effect of ghrelin in the course of ethanol-induced gastric ulcers in rats. J Physiol Pharmacol 2014; 65: 95-106.
- Warzecha Z, Ceranowicz D, Dembinski A, et al. Ghrelin accelerates the healing of cysteamine-induced duodenal ulcers in rats. Med Sci Monit 2012; 18: BR181-BR187.
- Warzecha Z, Kownacki P, Ceranowicz P, Dembinski M, Cieszkowski J, Dembinski A. Ghrelin accelerates the healing of oral ulcers in non-sialoadenectomized and sialoadenectomized rats. J Physiol Pharmacol 2013; 64: 657-668.
- Dembinski A, Warzecha Z, Ceranowicz P, et al. Ghrelin attenuates the development of acute pancreatitis in rats. J Physiol Pharmacol 2003; 54: 561-573.
- Dembinski A, Warzecha Z, Ceranowicz P, et al. Role of growth hormone and insulin-like growth factor-1 in the protective effect of ghrelin in ischemia/reperfusion-induced acute pancreatitis. Growth Horm IGF Res 2006; 16: 348-356.
- Warzecha Z, Ceranowicz P, Dembinski A, et al. Therapeutic effect of ghrelin in the course of cerulein-induced acute pancreatitis in rats. J Physiol Pharmacol 2010; 61: 419-427.
- Ceranowicz D, Warzecha Z, Dembinski A, et al. Role of hormonal axis, growth hormone - IGF-1, in the therapeutic effect of ghrelin in the course of cerulein-induced acute pancreatitis. J Physiol Pharmacol 2010; 61: 599-606.
- Bukowczan J, Warzecha Z, Ceranowicz P, Kusnierz-Cabala B, Tomaszewska R, Dembinski A. Therapeutic effect of ghrelin in the course of ischemia/reperfusion-induced acute pancreatitis. Curr Pharm Des 2015; 21: 2284-2290.
- Konturek PC, Brzozowski T, Engel M, et al. Ghrelin ameliorates colonic inflammation. Role of nitric oxide and sensory nerves. J Physiol Pharmacol 2009; 60: 41-47.
- Hosomi S, Oshitani N, Kamata N, et al. Phenotypical and functional study of ghrelin and its receptor in the pathogenesis of Crohn’s disease. Inflamm Bowel Dis 2008; 14: 1205-1213.
- Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis 2006; 12: 100-105.
- Peracchi M, Bardella MT, Caprioli F, et al. Circulating ghrelin levels in patients with inflammatory bowel disease. Gut 2006; 55: 432-433.
- Ates Y, Degertekin B, Erdil A, Yaman H, Dagalp K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig Dis Sci 2008; 53: 2215-2221.
- Gonzalez-Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology 2006; 130: 1707-1720.
- Pamukcu O, Kumral ZN, Ercan F, Yegen BC, Ertem D. Anti-inflammatory effect of obestatin and ghrelin in dextran sulfate sodium-induced colitis in rats. J Pediatr Gastroenterol Nutr 2013; 57: 211-218.
- De Smet B, Thijs T, Moechars D, et al. Endogenous and exogenous ghrelin enhance the colonic and gastric manifestations of dextran sodium sulphate-induced colitis in mice. Neurogastroenterol Motil 2009; 21: 59-70.
- Zhao D, Zhan Y, Zeng H, Moyer MP, Mantzoros CS, Pothoulakis C. Ghrelin stimulates interleukin-8 gene expression through protein kinase C-mediated NF-kappaB pathway in human colonic epithelial cells. J Cell Biochem 2006; 97: 1317-1327.
- Dembinski A, Warzecha Z, Ceranowicz P, et al. Role of capsaicin-sensitive nerves and histamine H1, H2, and H3 receptors in the gastroprotective effect of histamine against stress ulcers in rats. Eur J Pharmacol 2005; 508: 211-221.
- Warzecha Z, Dembinski A, Brzozowski T, et al. Gastroprotective effect of histamine and acid secretion on ammonia-induced gastric lesions in rats. Scand J Gastroenterol 2000; 35: 916-924.
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 1982; 78: 206-209.
- Dembinski A, Warzecha Z, Konturek SJ, et al. Extract of grapefruit-seed reduces acute pancreatitis induced by ischemia/reperfusion in rats: possible implication of tissue antioxidants. J Physiol Pharmacol 2004; 55: 811-821.
- Vilaseca J, Salas A, Guarner F, Rodriguez R, Martinez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis. Gut 1990; 31: 539-544.
- Hellmich MR, Evers BM. Regulation of gastrointestinal normal cell growth. In: Physiology of the Gastrointestinal Tract, Johnson LR (ed.) Amsterdam - Boston - Heildelberg, Elsevier Academic Press, 2006, pp. 435-458.
- Greant P, Delvaux G, Willems G. Influence of stress on epithelial cell proliferation in the gut mucosa of rats. Digestion 1988; 40: 212-218.
- Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci 1994; 107: 3569-3577.
- Konturek SJ, Brzozowski T, Dembinski A, Warzecha Z, Konturek PK, Yanaihara N. Interaction of growth hormone-releasing factor and somatostatin on ulcer healing and mucosal growth in rats: role of gastrin and epidermal growth factor. Digestion 1988; 41: 121-128.
- Konturek SJ, Dembinski A, Warzecha Z, Brzozowski T, Gregory H. Role of epidermal growth factor in healing of chronic gastroduodenal ulcers in rats. Gastroenterology 1988; 94: 1300-1307.
- Brzozowski T, Konturek PC, Konturek SJ, et al. Effect of local application of growth factors on gastric ulcer healing and mucosal expression of cyclooxygenase-1 and -2. Digestion 2001; 64: 15-29.
- Beckert S, Class N, Farrahi F, Coerper S. Growth hormone enhances gastric ulcer healing in rats. Med Sci Monit 2004; 10: BR255-BR258.
- Podolsky DK. Regulation of intestinal epithelial proliferation: a few answers, many questions. Am J Physiol 1993; 264: G179-G186.
- Sorbye H, Svanes K. The role of blood flow in gastric mucosal defence, damage and healing. Dig Dis 1994; 12: 305-317.
- Orlando RC. The integrity of the esophageal mucosa. Balance between offensive and defensive mechanisms. Best Pract Res Clin Gastroenterol 2010; 24: 873-882.
- Leung FW, Reedy TJ, Van Deventer GM, Guth PH. Reduction in index of oxygen saturation at margin of active duodenal ulcers may lead to slow healing. Dig Dis Sci 1989; 34: 417-423.
- Leung FW, Su KC, Pique JM, Thiefin G, Passaro E, Jr, Guth PH. Superior mesenteric artery is more important than inferior mesenteric artery in maintaining colonic mucosal perfusion and integrity in rats. Dig Dis Sci 1992; 37: 1329-1335.
- Warzecha W, Dembinski A, Brzozowski T, et al. Histamine in stress ulcer prophylaxis in rats. J Physiol Pharmacol 2001; 52: 407-421.
- West SD, Helmer KS, Chang LK, Cui Y, Greeley GH, Mercer DW. Cholecystokinin secretagogue-induced gastroprotection: role of nitric oxide and blood flow. Am J Physiol Gastrointest Liver Physiol 2003; 284: G399-G410.
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27: 519-550.
- Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity 2013; 39: 1003-1018.
- Norman J, Franz M, Messina J, et al. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery 1995; 117: 648-655.
- Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol 2011; 41: 1203-1217.
- Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012; 11: 633-652.
- De Koning HD, Schalkwijk J, van der Ven-Jongekrijg J, Stoffels M, van der Meer JW, Simon A. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann Rheum Dis 2013; 72: 1634-1638.
- Herlin T, Fiirgaard B, Bjerre M, et al. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis 2013; 72: 410-413.
- Mullane KM, Kraemer R, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods 1985; 14: 157-167.
- Barone FC, Hillegass LM, Price WJ, et al. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res 1991; 29: 336-345.
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol 2005; 77: 598-625.
- Romero FJ, Bosch-Morell F, Romero MJ, et al. Lipid peroxidation products and antioxidants in human disease. Environ Health Perspect 1998; 106 (Suppl. 5): 1229-1234.
- Kwiecien S, Jasnos K, Magierowski M, et al. Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress - induced gastric injury. J Physiol Pharmacol 2014; 65: 613-622.
- Parks DA. Oxygen radicals: mediators of gastrointestinal pathophysiology. Gut 1989; 30: 293-298.
- Pohle T, Brzozowski T, Becker JC, et al. Role of reactive oxygen metabolites in aspirin-induced gastric damage in humans: gastroprotection by vitamin C. Aliment Pharmacol Ther 2001; 15: 677-687.
- Kwiecien S, Pawlik MW, Brzozowski T, et al. Nitric oxide (NO)-releasing aspirin and (NO) donors in protection of gastric mucosa against stress. J Physiol Pharmacol 2008; 59 (Suppl. 2): 103-115.
- Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 2012; 237: 474-480.
- Evans MD, Cooke MS. Factors contributing to the outcome of oxidative damage to nucleic acids. BioEssays 2004; 26: 533-542.
A c c e p t e d : November 3, 2015