COMBINATION OF HYPERHOMOCYSTEINEMIA AND ISCHEMIC TOLERANCE
IN EXPERIMENTAL MODEL OF GLOBAL ISCHEMIA IN RATS
INTRODUCTION
Homocysteine (Hcy) is a toxic, sulfur-containing intermediate of methionine metabolism. Hyperhomocysteinemia (hHcy) as a consequence of impaired Hcy metabolism or defects in crucial co-factors that participate in its recycling, is assumed as an independent cerebrovascular risk factor (1-5).
Several potential mechanisms have been proposed to explain the possible connection between hHcy and central nervous system (CNS) disorders (6). Ischemic brain stroke is a heterogenous disorder with multiple and sequentional pathogenesis which eventually could lead to the neurodegenerative disorders. Earlier studies showed that Hcy, similar to stroke, induces oxidative stress in the brain through the activation of glutamate, N-methyl-D-aspartate (NMDA) type receptors, an autoxidation of Hcy and consequent reactive species generation (4, 5, 7). Hcy also reduces antioxidant defence and increases lipid peroxidation as well as an oxidative DNA damage in the brain (5, 6, 8-13). It has been demonstrated that homocysteine thiolactone-induced seizures in adult rats, which could co-occured after stroke, are aggravated by the inhibition of inducible nitric oxide synthase (14). Rodent studies in conditions of hHcy documented a direct detrimental effect to several key pathways, or changes in the cellular methylation potential (4, 15, 16). hHcy also interferes with the expression of molecules related to survival of vulnerable neurons (2).
The acute ischemic event follows the selective cell death of vulnerable pyramidal neurons of the hippocampal CA1 (Cornu Ammonis 1) region and neurons of the cerebral cortex and striatum (2, 17-21). Previous investigations had already shown that oxidative stress is an important common mechanism of brain injury in both pathologies (2, 22). In a series of studies from our laboratory (19, 23-25) we have found that ischemia-reperfusion (IR) injury initiates the time-dependent changes in activation of several proteins related to the neuronal death or survival. Ischemic preconditioning (IPC) manifests a powerful protective potential against ischemic cell death in selectively vulnerable neurons (2, 13, 17, 19, 23, 26-30) conducted in various animal models (31, 32) and in humans (12, 13, 23).
The ample evidence is documenting the role of a variety of signalling molecules in the IPC phenomenon (2, 12, 13, 19, 23, 24) including mitogen activated protein kinases (MAPKs; 10, 11, 33, 34). MAPKs are family of related serine/threonine kinases (11, 24, 25, 33, 35, 36) which are activated by growth factors, oxidative stress, increasing intracellular Ca2+ levels and glutamate receptor stimulation, all factors of which are activated by IR insult (24, 25, 36, 37) as well as by hHcy (10, 11, 33, 34, 37). Although many studies have been focused on determining the influence of hHcy in the brain (11, 38, 39), there is no information about the effect of hHcy on the ischemic tolerance mechanisms.
As we have shown in our previous researchs, IPC initiates a massive increase of MAPK/p-ERK (extracellular-signal-regulated kinase) in the rat cerebral cortex and hippocampus (24, 25). No previous study has examined the possible cross-talk between MAPK/p38 and MAPK/ERK in association with hHcy during IR injury and IPC. Therefore, the present study was undertaken to describe the extension of neurodegenerative changes and imbalance in MAPK/p-ERK1/2 and MAPK/p-p38 expression in the rat brains with induced hHcy following IPC. We used a model of experimental hHcy originally developed by Streck et al. (8), in which the Hcy levels were similar to those found in human homocystinuria (40).
MATERIALS AND METHODS
Evocation of ischemia-reperfusion and ischemic preconditioning
Animal studies were performed using protocol approved by the State Veterinary and Food Department of the Slovak Republic (no 727/12-221) in the grant under the title "Molecular mechanisms of neuroprotection and ischemic tolerance". Adult male Wistar rats (mean body weight of 320 g, total n = 120) were housed in a menagerie under the standard conditions with a temperature of 22 ± 2°C, and had periodical daylight variation at 12 h intervals. Food and water were provided ad libitum. Global forebrain ischemia was induced by using the standard 4-vessel occlusion model as described in previous works (2, 19, 24, 25). Briefly, on the day 1 both vertebral arteries were irreversibly occluded for 10 min by thermo coagulation through the alar foramina after anesthesia with 2.5% halothane in a mixture of oxygen/nitrous oxide (30/70%). No visible effect on the animals was observed. On the day 2, both common carotid arteries were occluded for 15 min by small atraumatic clips under the anesthesia as described from day 1. The halothane was removed from the mixture two minutes before carotid occlusion. Normothermic conditions (37°C) were monitored by a microthermistor placed in the ear and temperature was maintained using a homeothermic blanket. Sham control animals were prepared in the same way without carotid occlusion. The rats then underwent 15 min ischemia. Criteria for forebrain ischemia comprised loss of the righting reflex, mydriasis and paw extension. We used rats that became unresponsive, lost the righting reflex during bilateral carotid artery occlusion and showed no seizures during and after ischemia. Those that met the criteria for global forebrain ischemia were divided into the groups in the same way as non-treated animals. IPC was induced by 5 min of sublethal ischemia followed by 2 days of reperfusion. The rats then underwent lethal ischemia for 15 min as described above. We used 12 animals/group due to different preparation of biological material for biochemical (n = 6) and histological (n = 6) methods. After ischemia, animals were sacrificed by decapitation in a mild halothane anesthesia. Brains were dissected and processed immediately. Control animals for both ischemia group and preconditioned ischemia group underwent the same procedure with the exception of carotid occlusion.
Chemically-induced hyperhomocysteinemia
Homocysteine (Hcy) - (Sigma-Aldrich, Bratislava, Slovak Republic) was dissolved in 0.85% (w/v) NaCl solution (saline) and buffered to pH 7.4. Hcy solution (0.45 µmol/g body weight) was administered subcutaneously twice a day at 8 h intervals for 14 days. It is well known that Hcy crosses the blood/brain barrier and reaches the peak in the cerebrum and parietal cortex between 15 – 60 min after subcutaneous injection (8, 10). Doses of Hcy were calculated from pharmacokinetic parameters that were previously determined (8). Rats subjected to this treatment achieved plasma Hcy levels similar to those found in homocystinuric patients. (moderate hHcy; 40).
Experimental groups of animals
-
Groups of rats were randomized as follows:
- sham-operated control (naive) animals (C, n = 12);
- sham-operated hHcy control animals (Hcy-C, n = 12);
- the hHcy animals that underwent 15 min ischemia (Hcy-I) and 3, 24 and 72 h reperfusion (Hcy-IR-3 h rep, Hcy-IR-24 h rep and Hcy-IR-72 h rep, n = 12);
- the hHcy animals with induced IPC following 15 min ischemia (Hcy-IPC-I) and 3, 24 and 72 h reperfusion (Hcy-IPC-3 h rep, Hcy-IPC-24 h rep and Hcy-IPC-72 h rep, n = 12).
Determination of plasma homocysteine in animals
The animals have been executed 18 – 20 hours after the last Hcy injection. Their blood was collected in tubes containing EDTA (ethylenediaminetetraacetic acid). Blood was subsequently centrifuged and plasma was stored at –80°C. Total Hcy (tHcy) level in the plasma of animals was determined by enzymatic two part reagent system (Hcy liquid stable reagent kit, Erba Lachema, 50003526) using a chemical analyser (Siemens ADVIA 1650) at the Department of Clinical Biochemistry Comenius University and UNM in Martin, Slovakia.
Preparation of hippocampal homogenates
Hippocampi (n = 6/group) were homogenized in 10 mmol/l Hepes/KOH pH 7.4, 0.32 mol/l sucrose, 0.1 mmol/l phenylmethylsulfonyl fluoride and protease inhibitor cocktail solution (Roche). The homogenate was then centrifuged for 10 min at 1500 g. The pellet was discarded and the supernatant centrifuged for 45 min at 100,000g. The final pellet was resuspended in 10 mmol/l Hepes/KOH pH 7.4, 0.32 mol/l sucrose and stored at –80°C until further use.
Western blot analysis
Hippocampi from Sham-control, ischemic and pre-ischemic animals with/without Hcy treatment were homogenized as described above and resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After Western blot (WB) (Trans blot SD semi-dry transfer cell, Bio-rad) blots on nitrocellulose membrane were probed with rat polyclonal antibodies against MAPK/p-p38 (sc-101759, 1:300, Santa Cruz Biotechnology) as well as with mouse monoclonal antibodies against MAPK/p-ERK1/2 (sc-7383, 1:500, Santa Cruz Biotechnology) at room temperature for 3 hours. For normalization necessary to account the differences in total protein concentration between each sample, membranes were also probed with mouse monoclonal antibody against β-actin (sc-47778, 1:500, Santa Cruz Biotechnology) at room temperature for 3 hours. After washing with 0.05% phosphate-buffered saline (PBS)-Tween, the membranes were incubated with goat anti-mouse (MAPK/p-ERK1/2 and β-actin; sc-2005, 1:1000, Santa Cruz Biotechnology) and goat anti-rabbit (sc-2040, 1:1000, Santa Cruz Biotechnology) secondary antibodies conjugated with horseradish peroxidase for 1 hour and washed again with PBS-Tween. Finally, the membranes were developed using a Super Signal West Pico Chemiluminescence Substrate (ECL system from Pierce, #34080) and detected by Molecular Imager Gel Doc XR System (Bio-Rad) as described in (41). We analyzed six animals per experimental group. To reduce differences among rats, sample loading on SDS-PAGE and variability due to ECL detection, WBs were performed for each reperfusion time point per animal at least four times. Protein was measured using bovine serum albumin (BSA) as a standard described in (41).
Fluorescent immunohistochemistry
The above mentioned group of animals (n = 6/group) were anaesthetized with 2.5% halothane in a mixture of oxygen/nitrous oxide (30/70%) and perfused transcardially with 0.1 mol/l PBS (pH 7.4) followed by 4% paraformaldehyde in 0.1 mol/l PBS (pH 7.4; 24, 25). The brains were removed and postfixed with the same solution as above for 24 h at 4°C. The tissues were cryoprotected by infiltration using 30% sucrose for the next 24 hours at 4°C. The brain tissues were then frozen and sectioned with a cryostat at 30 µm, and the sections mounted into Superfrost Plus glass (Thermo scientific). Sections were permeabilized with 0.1% Triton X-100, pre-blocked with 10% BSA for 60 min. Primary antibodies used in the WB analysis have been mentioned previously (p-ERK and p-p38 MAPKs, each diluted 1:100). The tissue sections were incubated with O/N at 4°C using primary antibodies diluted in the 0.1% Triton X-100 solution with 10% BSA. Detection was performed using Alexa Fluor 488 goat-anti-mouse IgG (A11001, 1:100, Life Technologies) and Alexa Fluor 594 goat-anti-rabbit IgG (A11012, 1:100, Life Technologies)-conjugated secondary antibodies. Sections were mounted in Vectashield mounting medium containing 4’, 6-diamidino-2-phenylindole (DAPI; CA 94010, Vector Laboratories) according to the standard protocols. No immunoreactivity was detected in the absence of the primary antibodies. To reduce differences among rats, samples used for fluorescent immunohistochemical analysis were performed for each reperfusion time point per animal at least four times. Number of positive cells was accounted in the CA1 region of hippocampus and M1 (primary motoric brain) area of forebrain cortex by each animal at least in three different sections.
Fluoro-Jade C staining
Fluoro-Jade C was used as a marker for neurons undergoing degeneration. The sections mounted on the silanized glass were heated at 50°C for at least half an hour before staining. The slides were immersed in absolute alcohol for 3 min then 1 min in 70% alcohol and 1 min in distilled water. Then the slides were transferred to the solution of 0.06% potassium permanganate for 15 min and rinsed in distilled water for 2 min. After 120 min staining, they were rinsed 3 × 1 min in distilled water. The slides were dried at room temperature, cleared by xylene and cover slipped with Fluoromount™ Aqueous Mounting Medium (F4680, Sigma-Aldrich) according to the standard protocols.
In situ labelling of DNA fragmenattion by TUNEL (terminal transferase-mediated dUTP nick end labeling) assay
In situ nick end labelling of nuclear DNA fragmentation in the sections were performed in a humid chamber for 1 hour in the dark at 37°C with a TUNEL detection kit (In Situ Cell Death Detection Kit, Fluorescein, Roche) following the manufacturer's instructions. For each experiment, a positive control was prepared by treating the sections with 1 U µl–1 DNase I for 10 min at 37°C before labelling as above. The negative controls were labelled in parallel, except for the absence of the enzyme terminal deoxynucleotidyl transferase (TdT). The labelling reaction (TUNEL) was stopped with 2 × SSC (300 mmol/l NaCl, 30 mmol/l sodium citrate) and then the slides were rinsed with PBS (pH 7.2). The slides were counterstained with Fluoromount™ Aqueous Mounting Medium (F4680, Sigma-Aldrich), for fluorescence microscopy and kept in the dark at 4°C until microscopic observation was onducted.
Statistical analysis
Images of MAPK/p-p38, MAPK/p-ERK, Fluoro-Jade C and TUNEL immunoreactivity in the CA1 region of rat hippocampus as well as in the M1 region of rat forebrain were captured with an OLYMPUS fluoview FV10i confocal microscope in each animal. The brightness and contrast of each image file was uniformly calibrated using Adobe Photoshop version 2.4.1, followed by analysis using Image-Pro Plus 6.0 software. Values of the background staining were obtained and subtracted from the immunoreactive intensities. All results were presented as means ± S.E.M. ANOVA and Student-Neuman-Keuls tests were used to compare the control, IR and IPC groups with/without hHcy. The results from WB analysis were normalized to the control thus representing 100%. A value of P < 0.05 was considered to be statistically significant.
RESULTS
1. Determination of plasma homocysteine in animals
Determination of plasma Hcy in animals has shown that plasma tHcy level in naive controls with 14 days induced hHcy was significantly elevated as compared to the physiological level in the male Wistar rats (6.42 ± 1.65 µmol/l, 42) and reached 16.64 ± 1.38 µmol/l.
2. Acute histologic damage in the rat brain after induced hyperhomocysteine
2.1 Fluoro-Jade C
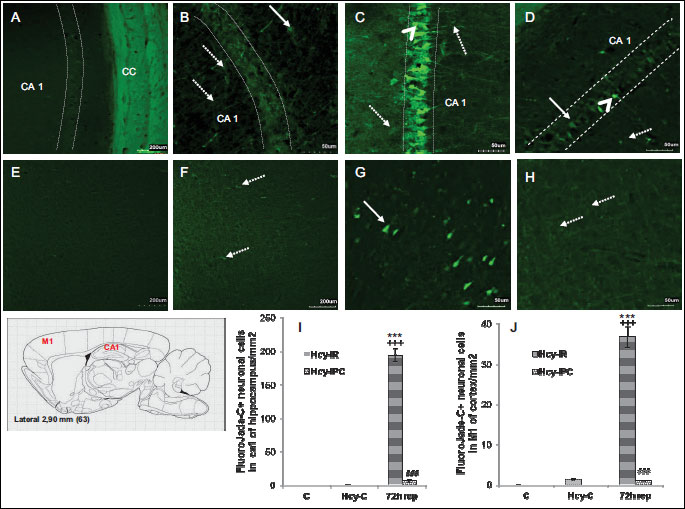
For demonstration of neuronal degeneration the tissue slices were analyzed using the Fluoro-Jade C in naive controls with/without induced hHcy as well as in the groups after IR insult and IPC 72 h after reperfusion. We detected none positive degenerating neurons in the hippocampi and M1 region in sham-controlled animals (Fig. 1A and 1E). In the naive control group with induced hHcy, the Fluoro-Jade C positive glial elements were localized mostly in stratum oriens of the CA1 region of hippocampus (Fig. 1B), suggesting activation of the astrocytes in spite of the weak afinity of Fluoro-Jade to glial elements (43, 44). The pattern of Fluoro-Jade C staining was similar in the extra-hippocampal regions (layers III-V of the cortical M1 region; Fig. 1F).
15 min ischemia followed by 72 h reperfusion with induced hHcy was capable of killing roughly 80 – 85% CA1 neurons in hippocampus (Fig. 1C). We found 647.7 times increase in the number of Fluoro-Jade C possitive cells in comparison to controls (194.7 ± 9.2; P < 0.001) and 149.49 times increase in comparison to naive controls with induced hHcy (1.3 ± 0.2; P < 0.001). Remarkably, IPC with induced hHcy decreased the number of degenerating cells in the CA1 region of hippocampus in comparison to Hcy-IR-72 h rep group. The number of Fluoro-Jade C+ cells in Hcy-IPC-72 h rep was decreased to 3.9% (P < 0.001; Fig. 1I), whereas there was no possitive labelling in naive control animals.
Interestingly, possitive cells were detected in the cortical M1 region in Hcy-C group. The number and density of Fluoro-Jade C+ cells was 122.4 times higher (36.7 ± 2.6; P < 0.001) in Hcy-IR-72h rep group in comparison to control (Fig. 1G) and 28.2 times higher (1.3 ± 0.1; P < 0.001) in comparison to the hHcy controls. On the other hand, the induced tolerance by IPC decreased the number of positive cells to 3% in Hcy-IPC-72 h rep group (Fig. 1H) (P < 0.001; Fig. 1J) in comparison to Hcy-IR-72 h rep group.
2.2 TUNEL assay
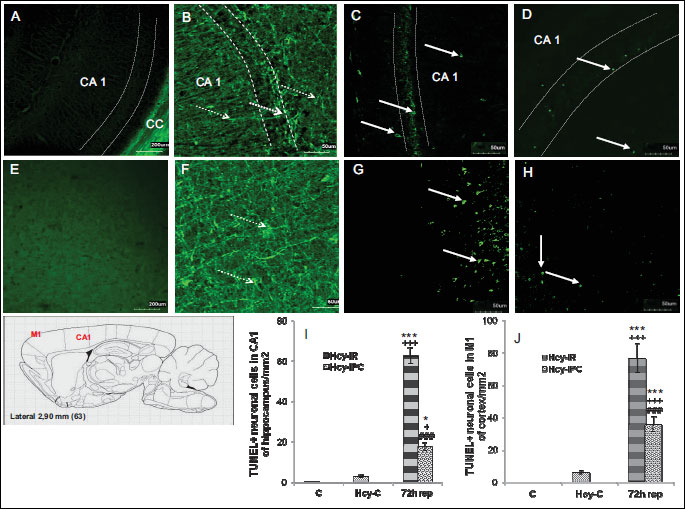
The poor TUNEL positivity was detected in control group in the CA1 of hippocampus as well as in M1 of forebrain cortex, (Fig. 2A and 2E). However, in hHCy controls was number of TUNEL possitive cells remarkably increased in both, the CA1 of hippocampus and M1 of cortex. The morphological features of TUNEL positive cells suggest for glial presence (Fig. 2B and 2F).
The number of TUNEL positive cells in the CA1 region of hippocampus in the Hcy-IR-72 h rep group (Fig. 2C) was 62.7 times (62.7 ± 4.05; P < 0.001) higher than in the control animals and 20.9 times (P < 0.001) higher compared to hHcy control (Fig 2I). On the contrary, in Hcy-IPC-72 h rep group, the number of TUNEL positive cells in the CA1 region of hippocampus was only slightly higher in comparison to the hHcy control (Fig. 2D) but reached 17.7 times (17.7 ± 2.03; P < 0.05) higher values than naive control group (Fig. 2I). Moreover, there was a significant difference between the Hcy-IPC-72 h rep group and controls with induced hHcy (5.9 times increase in the number of cells, P < 0.05). We found up to 28% significant decrease in the number of TUNEL positive neuronal cells in Hcy-IPC-72 h rep group in comparing to the Hcy-IR-72 h rep group (P < 0.001; Fig. 2I), suggesting a remarkable preventive role of IPC manoeuvre in the pyramidal CA1 neurons.
In the M1 sector of rat cortex (Fig. 2G and 2H) we detected 76.7 times increased number of TUNEL possitive cells in Hcy-IR-72 h rep group (76.7 ± 9.08; P < 0.001) in comparison to the controls and 12.78 times increased number (P < 0.001) of TUNEL positive cells in comparison to the hHcy control (Fig. 2J). In the Hcy-IPC-72 h rep group, the number of TUNEL positive cells reached 35.7 times higher quantity (35.7 ± 4.63; P < 0.001) in comparison to the naive controls and 5.95 times increased number (P < 0.001) in comparison to the hHcy control. IPC maneuver induced up to 47% significant decrease in number of TUNEL positive cells (P < 0.001) in the Hcy-IPC-72 h rep group in comparison to the Hcy-IR-72 h rep group, suggesting a remarkable preventive role of IPC for the pyramidal neurons in the cortical M1 area.
3. MAPK pathways in the hippocampal area after induced hyperhomocysteinemia
We first analyzed MAPK/p-ERK1/2 and MAPK/p-p38 proteins by Western blot analysis.
3.1 Western blot
This analysis clearly detected the presence of the both proteins in the injured area of the sham-operated controls and animals after the insult. Analysis was performed on the rat brain tissue. Comparing the expression of MAPK/p-ERK protein relate to the control group, we found a decreased level of MAPK/p-ERK to 76% (P < 0.01) in Hcy-I group and to 64% (P < 0.001) in Hcy-IR-24 h rep. As shown in the hHcy control, the protein level of MAPK/p-ERK1/2 (Fig. 3A) was increased to 188% (P < 0.001) in comparison to the controls. In the Hcy-IR groups we detected intensive descent in the expression (to 40%, P < 0.001), but to 63% increasing (P < 0.001) in comparison to the Hcy-IR-3 h rep and to 34% decreased expression (P < 0.001) in the Hcy-IR-24 h rep group. When compared I group, in the Hcy-IR-3 h rep we detected an increased expression of MAPK/p-ERK compared to the Hcy-I group (P < 0.001), however with the drop to 84% (P < 0.05) in the Hcy-IR-24 h rep group.
On the other hand, IPC initiated a general elevation of MAPK/p-ERK protein level in the CA1 hippocampal area. We detected a time dependent progressive elevation in the Hcy-IPC groups starting from the 195% (P < 0.001) increase at the ischemic time and reaching 232% (P < 0.001) in the Hcy-IPC-3 h rep and 273% (P < 0.001) in the Hcy-IPC-24 h rep, respectively. This elevation can be seen also by comparing the levels of hHcy control (to 124%. P < 0.001 in Hcy-IPC-3 h rep and to 145%, P < 0.001 in the Hcy-IPC-24 h rep). Interestingly, we found differences between the Hcy-IR and Hcy-IPC groups in the same period of reperfusion (Fig. 3A). IPC manoeuvre initiated the increase of MAPK/p-ERK to 257% (P < 0.001) compared to IR-I and this elevation was even higher at 3 h of reperfusion (up to 197%, P < 0.001) as well as at 24 h, where it reached 429% (P < 0.001).
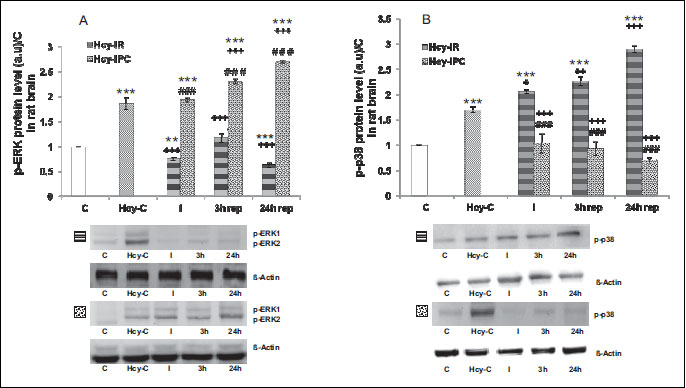
The highest level of activated MAPK/p38 protein was detected in the Hcy-IR groups. We found an increased level of MAPK/p-p38 also in the hHcy control to 170% (P < 0.001). In the Hcy-I group compared to controls, the level was elevated to 206% (P < 0.001) and further elevation was detected at higher reperfusion times to 227% (P < 0.001) in Hcy-IR at 3 h and to 290% (P < 0.001) in Hcy-IR at 24 h, respectively. The expression of MAPK/p-p38 protein was significantly elevatedin the Hcy-I group (to 121%, P < 0.05), in the Hcy-IR-3 h rep (to 133%, (P < 0.01) as well as in the Hcy-IR-24 h rep (to 171%, (P < 0.001) when compared to hHcy control. The protein level of MAPK/p-p38 was increased to 141% (P < 0.001) in the Hcy-I group compared to Hcy-IR-24 h rep (Fig. 3B).
Our previous studies suggested that IPC initiated the early response to the injury through the decreased levels of MAPK/p-p38 (24, 25). In the present experiments we detected significant differences between the individual groups. In the hHcy-IPC groups comparing to the hHcy controls were decreased the protein level to 61% (P < 0.001), to 55% (P < 0.001) in the Hcy-IPC at 3 h and to 41% (P < 0.001) in the Hcy-IPC-24 h rep group, respectively (Fig. 3B).
3.2 Double-staining immunoanalysis
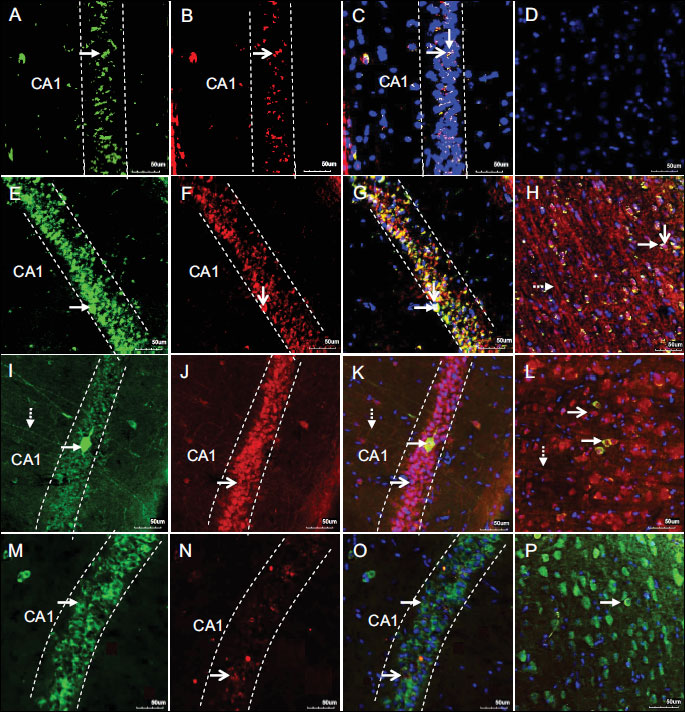
To further confirm the WB results we used a fluorescent immunohistochemistry method that detected immunoreactivity of MAPK/p-ERK1/2 and MAPK/p-p38 in the hippocampal area as well as in the cortical M1 region of rat forebrain. The figures show the representative pictures from the control, hHcy control, Hcy-IR-24 h rep and Hcy-IPC-24 h rep groups. Signals corresponding to MAPK/p-ERK1/2 and MAPK/p-p38 were predominantly located within the cytoplasm of perikarya and neuropil in histologically normal tissue (Fig. 4A-4D). The cytoplasmic immunoreactivity localization of MAPK/p-p38 as well as for MAPK/p-ERK was observed also in the CA1 area and M1 region of rat forebrain in hHcy controls (Fig. 4E-4H). As shown in Hcy-IR-24 h rep group (Fig. 4I-4L), the number of MAPK/p-p38+ cells was increased in the CA1 of hippocampus as well as in the M1 area of cortex. In contrast, immunoreactivity of MAPK/p-ERK 24 h after ischemia was decreased that is in the correlation with WB analysis. However, (Fig. 4M-4P) IPC induced a significant reduction of MAPK/p-p38 immunoreactivity. Preischemic manoeuvre had a contrary effect on MAPK/p-ERK protein expression when compared to the Hcy-IR-24 h rep group.
DISCUSSION
Mild to moderate hHcy is considered as an independent risk factor for stroke and neurodegenerative diseases (2, 10, 12, 13, 37). Several studies demonstrated that repeated doses of L-homocysteine (33) or even a single subcutaneous injection of 0.3 – 0.6 µmol/l Hcy induced significant cell death of cortical neurons. An in vitro investigations indicated that the treatment of hippocampal or cortical neurons with DL-Hcy (0.5 - 250 µmol/l) resulted in a dose-dependent increase in the extent of apoptosis (45). Although there is remarkable data on Hcy metabolism, the neuropathology linked with hHcy is not fully elucidated yet (12, 15). In our previous study we showed that hHcy had remarkable effect on mRNA SPCA (Secretory Pathway Ca2+-ATPase) expression, mainly in the cortical area (2).
Only limited number of experiments describe the mutual influence of co-morbid hHcy to ischemic damage on animal models of human stroke. Thus, incorporation of animal models more consistent with the clinical population afflicted by stroke are urgently needed for proper exploration of the disease´s etiology (12). Here, we suggest that hHcy alone (as an example of metabolic stress) and/or in combination with IR injury affects mechanisms of the ischemic tolerance induced by IPC manoeuvre. Prominent features of those changes were detected by using markers of cell damage/degeneration and changes in MAPKs expression in the CA1 hippocampal region and M1 cortical sector. We have previously shown that IR insult leads to the neurodegeneration of neurons in the CA1 region of hippocampus as detected by 75 – 80% Fluoro-Jade C+ and 90 times (9 ± 1.2 cells/mm2) higher TUNEL+ cells in comparison to the control, however only with minimal changes in cortex. On the other hand, IPC leads to supression of the number of positive cells and conferred neuroprotection (24, 25). Interestingly, the effect of homocysteine on cellular degeneration and following morphological changes was observed in the rat hippocampal and cortical regions. The increased number of Fluoro-Jade C+ and TUNEL+ neuronal and glial elements supports this effect of hHcy. The thickened and collapsed processes thatpoorly extend to the area of pyramidal neurons in CA1 region and M1 cortex are presumable due to the morphological alterations of astrocytes and cytoskeletal remodelling. This might suggest for the ensuing development of hHcy-associated neuronal cell damage (10, 33, 34, 37, 45-47). Astrocytes are highly plastic cells and their dynamic morphological changes could affect the intercellular communication with surrounding synapses that are important in the development of brain lesions (48). Maler et al. (49) reported that Hcy doses of 2 mmol/l and above induced a dose-dependent cytotoxic effect on cortical astrocytes. Astrocytes regulate expresion of the NMDA receptor subtypes, which increase neuronal sensitivity to glutamate toxicity and thus accelerate the initial step in a program of reactive astrogliosis and dynamics of the astrocyte response to damage (43, 44, 50). On the other hand, in response to the injury, astrocytes synthesize a number of factors that may play either neuroprotective or neurotoxic roles. At least in in vitro experiments with cultured cortical neurons, Loureiro et al. (47) describe their preventive role against the deleterious effect of Hcy.
In our previous studies, we have documented significant reductions of oxidative changes (51) and supression of cell degeneration in the hippocampus as well as in cortex of rat brain as an indicative consequence of preischemic treatment (2, 13, 24, 25). In this study we found that the combination of hHcy and IR insult followed by 72 h of reperfusion led to the extensive accumulation of damaged neurons in the CA1 and cortical regions. As we expected, IPC in the both paradigms (without and in hHCy condition) preserved the majority of neurons, as seen by decreasing the number of Fluoro-Jade C+ and TUNEL+ neuronal cells.
Recently, we have shown that IPC prior to lethal ischemia affects MAPK/ERK and MAPK/p38 pathways in the cerebral cortex (24) as well as in hippocampus (25). There is only sparse literature data focusing on the effect of Hcy on the MAPKs protein expression in neuronal cells (10, 11, 33). Our results suggest that the combination of both stressors (ischemia + Hcy) affects considerably the MAPKs pathways expression. hHcy-IR induces the MAPK/p38 expression as detected by WB and immunoanalysis. Poddar and Paul (11) showed a biphasic response of MAPK/p38 activation in the Hcy-NMDA induced neuronal damage in vitro, characterized by the initial rapid elevation followed by a delayed and more prolonged secondary increase, where the later peak was primarily involved in mediating of the Hcy-induced cell death. They also showed that this secondary activation of MAPK/p38 correlates with the upstream MAPK/ERK activation, which plays a role in facilitating of the Hcy-induced cell death. Our results are consonant with the conclusions of this study. Interestingly, we found that the MAPK/p38 pathway was activated also in hHcy control group. We previously reported that IR injury induces only slight increase of MAPK/p38 expression (24, 25). However, the combination of the both stressors (ischemia + hHcy) leads to the massive activation of MAPK/p-p38 with maximum at 24 h after reperfusion. This dynamic MAPK/p38 activation could contribute to more extensive progression of tissue injury (11, 35, 52, 53). Remarkably, the effect of the IPC manoeuvre on the decreasing of MAPK/p-p38 expression in ongoing reperfusion times indicates for proposed neuroprotective mechanism. The MAPK/p-p38 pathway was suppressed also by combination of two stressors
MAPK/ERK are versatile protein kinases that are ubiquitiously expressed in the CNS. We have already shown that in the hippocampus and cerebral cortex activated MAPK/ERK parallels neuroprotection induced by IPC (24, 25). The robust expression changes in hippocampus and modest post-translational changes in MAPK/ERK pathway in less sensitive vulnerable neurons of the cortical layers III and V (24, 25) corresponds with results of similar experimental models (34, 35, 53-55). Extensive studies have shown an interplay and tight integration of MAPK/ERK signalling in promoting neuronal cell death both in development and in neurodegenerative disorders (11, 33). It has been proposed that transient activation of MAPK/ERK kinase has different consequences as compared with sustained activation (11, 33, 56). Transient activation of MAPK/ERK plays a pivotal role in neuronal maturation, survival, and long-term potentiation (57). On the other hand, sustained activation of MAPK/ERK may play a critical role in triggering pro-apoptotic signals and neuronal cell death (11, 33). As documented in this study, the immunoreactivity of MAPK/p-ERK after IPC with induced hHcy was found in the early stages of reperfusion, with maximum level at 24 h, and its activation is probably associated with neuronal protection induced by IPC (34, 58-59). The slight activation of MAPK/ERK was detected also in the hHcy- control group. It is well known that hHcy mediates glutamate-mediated NMDA receptor stimulation, which eventually leads to the activation of both stimulatory and inhibitory pathways involved in the modulation of MAPK/ERK signaling (33). In fact, the dual role of MAKP/ERK kinases in cell survival and death suggests that a unique profile of gene expression may be elicited depending on the duration and/or magnitude of MAPK/ERK kinase activation (11). Thus, the duration of MAPK/ERK kinase activation following MAPK/p38 stimulation depends on the nature of the extracellular stimuli and may have different consequences on intracellular signalling pathways eventually leading to different cellular responses.
IPC could be potentially used as a preventive measure in a high risk individuals or as a precaution against secondary stroke following medical procedures such as aneurysm repair or cardiac surgery (12, 60). From the clinical point of view, IPC using may bring difficulties (12, 61). Initial pre-clinical studies of this phenomenon realized that pharmacological agents are also effective. However, there are some other agents already clinically used. In the case of naturally occurring human strokes which can not be predicted, manouevre of postconditioning may be a therapeutic strategy that could be used afterward to accelerate or enhance protective mechanisms or as a precaution against stroke recurrence (62). In correlation with our findings, some evidence suggests that although preconditioning may be beneficial in the short term, long term structural changes in the brain indicate that tissue damage is merely being postponed (12, 60). Clinical studies are needed to test the safety and efficacy of these novel strategies in humans (61).
In conclusion, we have shown that hHcy is associated with a selective degeneration of cortical and limbic structure including hippocampal area. The degeneration involves loss of neurons, glial growth, hypertrophy of astrocytes and probably sprouting of new connections. The present morphological findings indicate the astrocytes are the first neural cells participating in the deleterious actions of Hcy on the CNS. Apparently, astrocytes are able to respond to mild hHcyby reorganizing their cytoskeleton, surviving and protecting neurons from the damage (47).
Moreover, our study suggests that there are at least two different ways in which neuronal tissue responds to hHcy. Induction of hHcy alone leads to neuronal cell death and morphological changes in the hippocampus and cortex that corresponds with findings of previous reported studies (2, 10, 11, 33, 47). Combination of hHcy with more intense stimuli (ischemia) causes more prominent changes in cortex than in hippocampus. Importantly, IPC manoeuvre, even if combined with hHcy, still preserves the neuronal tissue from lethal ischemic effect. The other important findings arosen from this study is that MAPK/p38 promotes neuronal cell death, whereas MAPK/ERK activation opposes apoptosis. Finally, the study provides evidence for the interplay and tight integration between ERK and p38 MAPKs signaling mechanism in response to mild hHcy and also in association with IR insult/IPC challenge in rat brain.
We report that preischemia had a protective/stimulatory influence on the MAPK/ERK protein activation under both stress conditions (naive ischemia and/or hHcy- ischemia). This study also emphasizes the opposing effect of MAPK/ERK and MAPK/p38 on cell survival and cell death in hHcy and its combination with IPC challenge at least in rat model of human stroke. We hope that identification of the effects of co-morbid factors in the mechanisms of ischemic tolerance would lead to improved therapeutics, especially in the brain tissue. Further evaluation of this molecular pathway is necessary to establish the role of hHcy in the progression of neurological disorders.
Acknowledgements: The authors are grateful to Mrs. Zdenka Cetlova and Mr. Jan Fillo for their excellent help with animals.
This study was supported by Grants VEGA 00213/12, 00229/15 and from the Ministry of Education of the Slovak Republic, Ministry of Health of the Slovak Republic and by project "Identification of novel markers in diagnostic panel of neurological diseases" code: 26220220114, co-financed from EU sources and European Regional Development Fund.
Conflict of interests: None declared.
REFFERENCES
- Rabaneda LG, Carrasco M, Lopez-Toledano MA, et al. Homocysteine inhibits proliferation of neuronal precursors in the mouse adult brain by impairing the bFGF signaling cascade and reducing Erk1/2-dependent cyclin E expression. FASEB J 2008; 22: 3823-3835.
- Pavlikova M, Kovalska M, Tatarkova Z, Sivonova-Kmetova M, Kaplan P, Lehotsky J. Response of secretory pathways Ca2+ ATPase gene expression to hyperhomocysteinemia and/or ischemic preconditioning in rat cerebral cortex and hippocampus. Gen Physiol Biophys 2011; 30 (Spec.): S61-S69.
- Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice Am J Physiol Heart Circ Physiol 2006; 290: H1206-H1213.
- Sachdev PS. Homocysteine and brain atrophy. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 1152-1161.
- Sharma VK. Homocysteine induced dementia: collecting evideneces for Alzheimer's disease. Int J Pharma Biol Sci 2010; 1: 1-14.
- Zhuo JM, Wang H, Pratico D. Is hyperhomocysteinemia an Alzheimer's disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci 2011; 32: 562-571.
- Dorszewska J, Florczak-Wyspianska J, Oczkowska A, Dezor M, Prendecki M, Kozubski W. Homocysteine and asymmetric dimethylarginine concentrations in the plasma of Alzheimer's disease patients with varying degrees of dementia. Adv Alzheimer Dis 2013; 2: 1-6.
- Streck EL, Matte C, Vieira PS, et al. Reduction of Na+,K+-ATPase activity in hippocampus of rats subjected to chemically-induced hyperhomocysteinemia. Neurochem Res 2002; 27: 1585-1590.
- Wyse AT, Zugno AI, Streck EL, et al. Inhibition of Na(+), K(+)-ATPase activity in hippocampus of rats subjected to acute administration of homocysteine is prevented by vitamins E and C treatment. Neurochem Res 2002; 27: 1685-1689.
- Matte C, Mussulini BH, dos Santos TM, et al. Hyperhomocysteinemia reduces glutamate uptake in parietal cortex of rats. Int J Dev Neurosci 2010; 28: 183-187.
- Poddar R, Paul S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J Neurochem 2013; 124: 558-570.
- Lehotsky J, Petras M, Kovalska M, Tothova B, Drgova H, Kaplan P. Mechanisms involved in the ischemic tolerance in brain: effect of the homocysteine. Cell Mol Neurobiol 2015; 35: 7-15.
- Petras M, Tatarkova Z, Kovalska M, et al. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J Physiol Pharmacol 2014; 65: 15-23.
- Hrncic D, Rasic-Markovic A, Macut D, Susic V, Djuric D, Stanojlovic O. Homocysteine thiolactone-induced seizures in adult rats are aggravated by inhibition of inducible nitric oxide synthase. Hum Exp Toxicol 2014; 33: 496-503.
- Serdarevic N, Begic L, Mulaomerovic-Softic A. The concentration of homocysteine in patients after ischemic brain stroke and vascular dementia. J Health Sci 2011; 1: 4-9.
- Abushik PA, Niittykoski M, Giniatullina R, et al. The role of NMDA and mGluR5 receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J Neurochem 2014; 129: 264-274.
- Sakata H, Niizuma K, Yoshioka H, et al. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci 2012; 32: 3462-3473.
- Liu XQ, Sheng R, Qin ZH. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol Sin 2009; 30: 1071-1080.
- Pavlikova M, Tatarkova Z, Sivonova M, Kaplan P, Krizanova O, Lehotsky J. Alterations induced by ischemic preconditioning on secretory pathways Ca2+-ATPase (SPCA) gene expression and oxidative damage after global cerebral ischemia/reperfusion in rats. Cell Mol Neurobiol 2009; 29: 909-916.
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nature Neurosci 2003; 6: 34-42.
- Zhao H, Joo S, Xie W, Ji X. Using hormetic strategies to improve ischemic preconditioning and postconditioning against stroke. Int J Physiol Pathophysiol Pharmacol 2013; 5: 61-72.
- Ataie A, Sabetkasaei M, Haghparast A, Hajizadeh Moghaddam A, Ataie R, Nasiraei Moghaddam Sh. An investigation of the neuroprotective effects of curcumin in a model of homocysteine - induced oxidative stress in the rat's brain. Daru 2010; 18: 128-136.
- Lehotsky J, Burda J, Danielisova V, Gottlieb M, Kaplan P, Saniova B. Ischemic tolerance: the mechanisms of neuroprotective strategy. Anat Rec (Hoboken) 2009; 292: 2002-2012.
- Kovalska M, Kovalska L, Pavlikova M, et al. Intracellular signaling MAPK pathway after cerebral ischemia-reperfusion injury. Neurochem Res 2012; 37: 1568-1577.
- Kovalska M, Kovalska L, Mikuskova K, Adamkov M, Tatarkova Z, Lehotsky J. p-ERK involvement in the neuroprotection exerted by ischemic preconditioning in rat hippocampus subjected to four vessel occlusion. J Physiol Pharmacol 2014; 65: 767-776.
- Lehotsky J, Racay P, Pavlikova M, et al. Cross-talk of intracellular calcium stores in the response to neuronal ischemia and ischemic tolerance. Gen Physiol Biophys 2009; 28: 104-113.
- Pignataro G, Scorziello A, Di Renzo G, Annunziato L. Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 2009; 276: 46-57.
- Zhao L, Liu X, Liang J, et al. Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res 2013; 1503: 78-88.
- Ding ZM, Wu B, Zhang WQ, et al. Neuroprotective effects of ischemic preconditioning and postconditioning on global brain ischemia in rats through the same effect on inhibition of apoptosis. Int J Mol Sci 2012; 13: 6089-6101.
- Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nature Neurosci 2011; 14: 1363-1368.
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 2006; 7: 437-448.
- Danielisova V, Burda J, Nemethova M, Gottlieb M, Burda R. An effective combination of two different method of postconditioning. Neurochem Res 2012; 37: 2085-2091.
- Poddar R, Paul S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J Neurochem 2009; 110: 1095-1106.
- Zhang QG, Wang RM, Han D, Yang LC, Li J, Brann DW. Preconditioning neuroprotection in global cerebral ischemia involves NMDA receptor-mediated ERK-JNK3 crosstalk. Neurosci Res 2009; 63: 205-212.
- Cargnello M, Roux PP. Activation and function of MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 2011; 75: 50-83.
- Lee CH, Yoo KY, Park OK, et al. Phosphorylated extracellular signal-regulated kinase 1/2 immunoreactivity and its protein levels in the gerbil hippocampus during normal aging. Mol Cells 2010; 29: 373-378.
- Carletti R, Tacconi S, Bettini E, Ferraguiti F. Stress activated protein kinases, a novel family of mitogen-activated protein kinases are heterogeneously expressed in the adult rat brain and differentially disturbed extracellular-signal-regulated protein kinases. Neuroscience 1995; 69: 1103-1110.
- Vizzardi E, Bonadei I, Zanini G, et al. Homocysteine and heart failure: an overview. Recent Pat Cardiovasc Drug Discov 2009; 4: 15-21.
- Menon V, Wang X, Greene T, et al. Homocysteine in chronic kidney disease: effect of low protein diet and repletion with B vitamins. Kidney Int 2005; 67: 1539-1546.
- Mudd SH, Levy HL, Skovby F. Disorders of transsulfuration. In: The Metabolic and Molecular Basis of Inherited Disease. CR Scriver, AL Beaudet, WS Sly, D Valle (eds). New York, McGraw-Hill, 2001, pp. 1279-1327.
- Tatarkova Z, Engler I, Calkovska A, et al. Effect of long-term normobaric hypoxia on oxidative stress in mitochondria of the guinea pig brain. Neurochem Res 2011; 36: 1475-1481.
- Martins PJ, Galdieri LC, Souza FG, et al. Physiological variation in plasma total homocysteineconcentrations in rats. Life Sci 2005; 76: 2621-2629.
- Colombo JA, Puissant VI. Fluoro Jade Stains early and reactive astroglia in the primate cerebral cortex. J Histochem Cytochem 2002; 50: 1135-1137.
- Anderson KJ, Fugaccia I, Scheff SW. Fluoro-jade B stains quiescent and reactive astrocytes in the rodent spinal cord. J Neurotrauma 2003; 20: 1223-1231.
- Kruman II, Culmsee C, Chan SL, et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci 2000; 20: 6920-6926.
- Kim SJ, Lee BH, Kim YM, Kim GH, Yoo HW. Congenital MTHFR deficiency causing early-onset cerebral stroke in a case homozygous for MTHFR thermolabile variant. Metab Brain Dis 2013; 28: 519-522.
- Loureiro SO, Romao L, Alves T, et al. Homocysteine induces cytoskeletal remodeling and production of reactive oxygen species in cultured cortical astrocytes. Brain Res 2010; 1355: 151-164.
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 2005; 6: 626-640.
- Maler JM, Seifert W, Huther G, et al. Homocysteine induces cell death of rat astrocytes in vitro. Neurosci Lett 2003; 347: 85-88.
- Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 2010; 79: 77-89.
- Lehotsky J, Urban P, Pavlikova M, Tatarkova Z, Kaminska B, Kaplan P. Molecular mechanisms leading to neuroprotection/ischemic tolerance. Effect of preconditioning on the stress reaction of endoplasmic reticulum. Cell Mol Neurobiol 2009; 29: 917-925.
- Lu Q, Rau TF, Harris V, Johnson M, Poulsen DJ, Black SM. Increased p38 mitogen-activated protein kinase signaling is involved in the oxidative stress associated with oxygen and glucose deprivation in neonatal hippocampal slice cultures. Eur J Neurosci 2011; 34: 1093-1101.
- Cao Q, Qian M, Wang XF, et al. Negative feedback regulation of Raf/MEK/ERK cascade after sublethal cerebra ischemia in the rat hippocampus. Neurochem Res 2011; 36: 153-162.
- Liu AJ, Hu YY, Li WB, Xu J, Zhang M. Cerebral ischemic pre-conditioning enhances the binding characteristics and glutamate uptake of glial glutamate transporter-1 in hippocampal CA1 subfield of rats. J Neurochem 2011; 119: 202-209.
- Lennmyr F, Karlsson S, Gerwins P, Ata KA, Terent A. Activation of mitogen-activated protein kinases in experimental cerebral ischemia. Acta Neurol Scand 2002; 106: 333-340.
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther 2006; 319: 991-997.
- Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem 2004; 11: 172-178.
- Gu Z, Jiang Q, Zhang G. Extracellular signal-regulated kinase 1/2 activation in hippocampus after cerebral ischemia may not interfere with postischemic cell death. Brain Res 2001; 901: 79-84.
- Yamashita S, Hirata T, Mizukami Y, et al. Repeated preconditioning with hyperbaric oxygen induces neuroprotection against forebrain ischemia via suppression of p38 mitogen activated protein kinase. Brain Res 2009; 1301: 171-179.
- Thompson JW, Dave KR, Young JI, Perez-Pinzon MA. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics 2013; 10: 789-797.
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 2009; 8: 398-412.
- Danielisova V, Gottlieb M, Bonova P, Nemethova M, Burda J. Bradykinin postconditioning ameliorates focal cerebral ischemia in the rat. Neurochem Int 2014; 72: 22-29.
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. London, Academic Press, 2006. http://www.scribd.com/doc/22822097/Rat-Brain-Atlas. Acessed 26 June 2012
A c c e p t e d : October 15, 2015