EFFECT OF ECONAZOLE AND BENZYDAMINE
ON SENSORY NEURONS IN CULTURE
2Angelini Research Center, Angelini S.p.A., S. Palomba-Pomezia (Rome), Italy
INTRODUCTION
Econazole is an imidazole derivative with wide anti-fungal activity that is largely used for the treatment of skin fungal infections such as athlete’s foot, tinea, pityriasis versicolor, ringworm, and jock itch (1-5). In addition, it is also used to treat vaginal infections, particularly those produced by Candida albicans (2, 6). Topical application of econazole creams is generally well tolerated although adverse effects are observed in 5% of patients using it for skin conditions, especially when used in the inguinal area, and intravaginally (2, 7, 8). The most common side effect is a burning sensation, along with itching and erythema (9, 10). These sensations can be augmented if neurogenic inflammation accompanies mycoses, as changes in gene expression sensitize nociceptors (11). Conversely, azole-mediated side effects can be attenuated by using local anesthetics and anti-inflammatory drugs that reduce the azole side effects (12-14). Benzydamine is a topical anti-inflammatory drug widely used for the treatment of mucosal inflammatory disorders (14). The pharmacological activity of benzydamine is mainly related to inhibition of proinflammatory cytokine synthesis (13), although it also displays anesthetic activity suggesting an action modulating neuronal excitability.
Topically, econazole nitrate is applied at 1% either as a cream or powder formulation. Permeation studies show that 10% of the applied drug permeates through the stratum corneum reaching the epidermis and dermis and a systemic distribution (15, 16). Epidermal econazole may be the responsible of the adverse effects complained by some patients (1, 17), as it can affect the cutaneous neuro-immune system. Indeed, the burning sensation may be driven by an effect of the anti-mycotic drug on nociceptor peripheral terminals promoting the firing of action potentials in these otherwise silent sensory neurons. Similarly, the itching perception may be mediated by release of pruritogenics from mast cells or by activating pruritogenic sensory neurons. Intriguingly, the effect of econazole on nociceptor excitability has not been assayed yet. Here, we have addressed this important question and investigated the consequences of exposing primary nociceptor cultures to micromolar concentrations of econazole. We found that econazole significantly augmented the firing of action potentials in nociceptors in culture. Furthermore, this activity was potentiated by pre-exposure of nociceptors to a pro-inflammatory mediator. Econazole raised intracellular Ca2+ by a complex mechanism that involved Ca2+ entry from the external milieu and release from the endoplasmic reticulum. Notably, econazole-induced cytosolic Ca2+ increases were not mediated by activation of a thermoTRP channel, thus implying the stimulation of another type of Ca2+-permeable channel. We also found that benzydamine binds to site 2 Na+ channel and it is an antagonist of voltage-gated Na+ channels expressed in nociceptors. As a result, benzydamine attenuated the nociceptor excitability evoked by capsaicin as well as that elicited by econazole in sensitized sensory neurons.
MATERIALS AND METHODS
Chemicals
Econazole nitrate and benzydamine chlorhydrate were obtained from Angelini S.p.A. Econazole was freshly prepared at a 200 mM stock concentration in DMSO. Benzydamine was freshly prepared at a 50 mM stock concentration in H2O. Capsaicin was dissolved in DMSO at a stock concentration of 10 mM (sigma). ATP was prepared in water at stock concentration of 10 mM (Sigma). Thapsigargin was dissolved in DMSO at stock concentration of 1 mM (Sigma). Clotrimazole [1-(chloro-a,a-diphenylbenzyl)-imidazole] was dissolved in DMSO at stock concentration of 10 mM (Sigma).
Binding assay to site 2 Na+ channel
Binding was performed using rat cerebral cortex homogenates with 10 nM [3H] batrachotoxin used as specific radioligand. Veratridine 300 µM was used to determine the non specific binding. Incubation was performed for 60 minutes at 22°C. Benzydamine was dissolved in DMSO as 10–2 M stock solution, further diluted in water and tested in duplicate at seven concentrations ranging from 0.01 µM to 100 µM to obtain competition curves. Results are expressed as a percent of control specific binding. Non linear regression analysis was performed on percent of inhibition of specific binding (GraphPad PRISM software).
Primary culture of sensory neurons
Primary cultures of rat nociceptors were obtained as described (18, 19). Briefly, dorsal root ganglia (DRG) were harvested from neonatal Wistar rats (3 – 5 days old). Ganglia were digested with 0.25% (w/v) collagenase (type IA) in DMEM-glutamax (Invitrogen) with 1% penicillin-streptomycin (P/S; 5000 U/ml, Invitrogen) for 1 h (37°C, 5% CO2). After digestion, rat DRG were mechanically dissociated using a glass Pasteur pipette. Single cell suspension was passed through a 100 µm cell strainer, and washed with DMEM-glutamax plus 10% foetal bovine serum (FBS; Invitrogen) and 1% P/S. Cells were seeded in DMEM-glutamax, 10% FBS and 1% P/S, supplemented with mouse 2.5S NGF 50 ng/mL (Promega), and 1.25 µg/mL cytosine arabinoside when required (37°C, 5% CO2).
All experiments were made 48 hours after cell seeding.
Calcium-imaging
Ca2+ microfluorography was carried out as previously described (18, 19). DRG on coverslips loaded with 5 µM fluo-4-acetoxymethyl ester plus 0.02% pluronic acid (Molecular Probes, Invitrogen) in HBSS extracellular solution (in mM): 140 NaCl, 4 KCl, 1 MgCl2, 1.8 CaCl2, 5 D-glucose, and 10 HEPES, pH 7.4 for 1 h (37°C, 5% CO2)) were mounted in an imaging chamber (RC-25, Harvard Apparatus), and continuously perfused with HBSS buffer or test solutions at room temperature (RT). For 0Ca2+ experiments the extracellular solution was (in mM): 140 NaCl, 4 KCl, 2.8 MgCl2, 0.5 EGTA, 5 D-glucose, and 10 HEPES, pH 7.4. Fluorescence from individual neurons was monitored through a 10 × air objective (Axiovert 200 inverted microscope, Carl Zeiss) with an ORCA-ER CCD camera (Hamamatsu Photonics). Protocols were pre-programmed and applied via computer-controlled pinch valves (Bioscience Tools, 5 mL/min flux). Fluo-4AM was excited at 500 nm using a computer-controlled lambda-10-2-filter wheel (Sutter Instruments) and emitted fluorescence was filtered at 535 nm. Images were acquired each 5 s with 100 ms exposure, and processed with AquaCosmos package software (Hamamatsu Photonics). Neuronal activity was evoked as indicated.
Patch clamp recordings
Whole-cell voltage clamp was made in SH-SY5Y-TRPV1 cells and primary cultures of rat nociceptors seeded on coverslips (19, 20), placed in RC-25 perfusion and connected to an external perfusion system at ~22°C. External solution contained (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 5 glucose, 20 mannitol, pH 7.4 (adjusted with NaOH). Internal pipette solution contained (in mM): 144 KCl, 2 MgCl2, 10 HEPES, 5 EGTA, pH 7.2 (adjusted with KOH). On sensory neurons, KCl was substituted by CsCl to block K currents. Membrane currents were acquired by using EPC10 HEKA Patch amplifier (HEKA Electronics). Cells were held at a resting membrane potential of –60 mV and at a sampling rate of 2.5 Hz. Patch glass pipettes with O.D 1.5 mm × I.D. 1.17 mm (Harvard Instruments) were made with a Sutter Pippete puller Equipment (Sutter Instruments) to have resistance of 2.5 – 6 MW. PatchMaster software (HEKA Electronics) was used for data acquisition and offline analysis. Cells were visually identified through × 20 air objective (Axiovert 200 inverted microscope, Carl Zeiss). TRPV1 was recorded from cells held at –60mV for 180 s in presence of 10 or 50 µM econazole, 5 µM clotrimazole and 500 nM capsaicin. The series resistance was usually < 10 MW and, to minimize voltage errors, was compensated to 50 – 80%. Sodium channel recordings were carried out in the whole cell configuration. Neurons were held at –60 mV and depolarized with a family of 50 ms step potentials from –100 mV to +40 mV in steps of 20 mV. The I – V curves were obtained by plotting the peak current as a function of the membrane potential. Recordings were carried out in the absence and presence of benzydamine as indicated.
Microelectrode array (MEA)
Cells were seeded on microelectrode array chambers previously coated with poly-L-lysine (8.33 µg/ml) and laminin (5 µg/ml). After 2 h, medium was replaced with DMEM-glutamax, 10% FBS and 1% P/S, supplemented with mouse 2.5S NGF 50 ng/ml (Promega), and 1.25 µg/mL cytosine arabinoside when required (37°C, 5% CO2). All experiments were made 48 h after cell seeding. Extracellular recordings were made using multiple electrode planar arrays of 60-electrode thin MEA chips, with 30 µm diameter electrodes and 200 µm inter-electrode spacing with an integrated reference electrode (Multichannel Systems GmbH) (19). The electrical activity of primary sensory neurons was recorded by the MEA1060 System at a sampling rate of 25 kHz (Multi Channel Systems GmbH, http://www.multichannelsystems.com), and analysed with the MC_Rack software version 4.3.0. TRPV1-mediated neuronal firing activity was evoked by 15 s applications of 0.5 µM capsaicin, using a continuous perfusion system (2 ml/min flux). Different concentrations (10, 50, 100 µM) of econazole were perfused to identify evoked neuronal spikes on cultured rat DRG neurons. Benzydamine at 50 µM and 100 µM concentration was used to study the blockade of TRPV1- and econazole-evoked neuronal spikes. Data were analysed using MC_RACK spike sorter and Neuroexplorer Software (Nex Technologies). An evoked spike was defined when the amplitude of the neuronal electrical activity overcame a threshold set at –20 µV. The recorded signals were then processed to extract mean spike frequency.
RESULTS
Econazole augments intracellular Ca2+ in nociceptors
Because econazole has been described to modulate Ca2+ homeostasis in different cell types (21-25), we evaluated the effect of econazole on intracellular Ca2+ levels in nociceptors in culture. First, we determined the maximal concentration of the azole that could be used without affecting cell viability. This is critical as econazole is insoluble in aqueous buffers and requires the use of DMSO. Cytotoxicity experiments in sensory neurons revealed that econazole at 50 µM for 24 h produced a 40% cell death, while at 100 µM the cell death was ł85%. Thus, to test in vitro an econazole concentration close to the clinically used, we selected 50 µM of the azole containing 0.025% DMSO, a concentration lower than that reporting a marginal inhibitory effect of neuronal activity (26). Indeed, this DMSO concentration did not significantly altered the cellular viability nor the neuronal activity of our cultures (data not shown).
As depicted in Fig. 1A, exposure of Fluo4-loaded nociceptors to 50 µM econazole evoked a rise in the Ca2+ signal indicating an increment in the intracellular Ca2+ concentration. This increase in cytosolic Ca2+ was half that evoked by 0.5 µM capsaicin and 100 µM allyl isothiocyanate (AITC) in the same neuron (Fig. 1A and 1B), but was significantly larger than that caused by 5 µM clotrimazole, an imidazole derivative that has been described as an agonist of the TRPV1 channel (27). Econazole-induced Ca2+ responses were present in virtually all types of nociceptors, independent of the type of channels expressed. For instance, econazole responses were observed in sensory neurons that do not express TRPA1 channels (Fig. 1A).
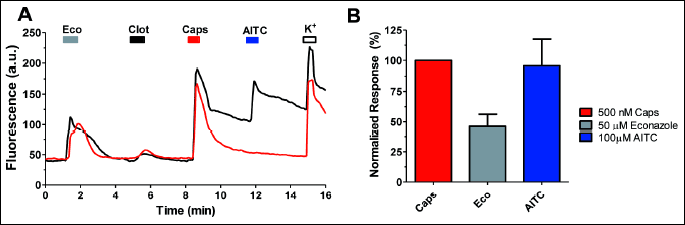
The increment of intracellular Ca2+ evoked by econazole raises the question as to whether it is mediated by activation of TRPV1, akin to clotrimazole. To address this question, we investigated the effect of econazole on TRPV1 channel activity in SH-SY5Y human neuroblastoma cells stably expressing this thermoTRP channel. These measurements were performed using patch-clamp. Exposure to capsaicin of voltage-clamp cells at –60 mV evoked a desensitizing inward current (Fig. 2A). Clotrimazole at 5 µM also evoked a desensitizing inward current, although of significantly lower amplitude than capsaicin, consistent with results by other groups (24). In marked contrast, 50 µM econazole did not elicit a detectable ionic current in these cells, indicating that this imidazole derivative is not an agonist of TRPV1. In addition, because econazole could evoke Ca2+ fluxes in nociceptors that do not express TRPA1 (Fig. 1A), we conclude that it was not an agonist of this thermoTRP. Therefore, the increment in intracellular Ca2+ evoked by econazole is likely mediated by another membrane-expressed channel or by release from intracellular stores.
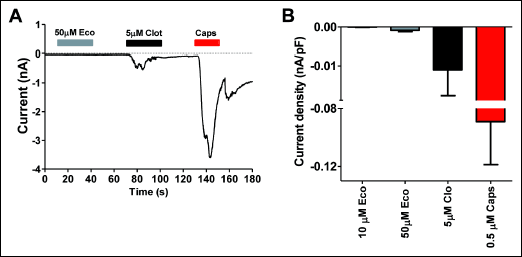 |
Fig. 2. Econazole does not activate TRPV1 channels. (A) Whole cell ionic currents activated by 50 µM econazole (Eco), 5 µM clotrimazole (Clot) and 0.5 µM capsaicin (Caps) from SH-SY5Y-TRPV1 cells, held at –60 mV. (B) Current density of inward currents elicited by Eco, Clot, and Caps at the concentrations depicted. Data are given as mean ± S.E.M., with n ł 7 (number of cells) and n = 3, number of cell cultures. |
Econazole releases Ca2+ from the endoplasmic reticulum
To understand the origin of the econazole-induced intracellular Ca2+ increases, we compared its activity in the presence and absence of extracellular Ca2+. Fig. 3A shows that econazole evoked rapid Ca2+ influx in control conditions (2 mM Ca2+ extracellular). Repetitive pulses of this imidazole derivative produced similar Ca2+ responses. Similarly, econazole was also able to produce an intracellular Ca2+ increase when the extracellular divalent cation was removed (Fig. 3B). Under these conditions, the second pulse exhibited a lower intensity than the first, consistent with the emptiness of an intracellular store. The number of responsive neurons in both conditions was identical, indicating that both mechanisms of regulating cytosolic Ca2+ coexist in the same sensory neurons (Fig. 3C).
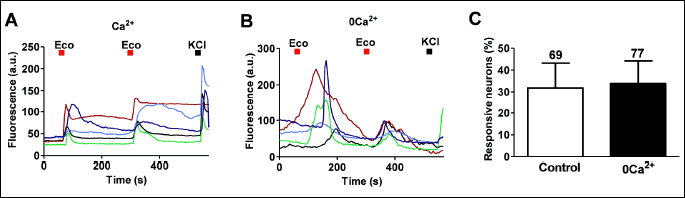
We next investigated the effect of releasing Ca2+ from the endoplasmic reticulum (ER) on the econazole-induced rise in cytosolic Ca2+. In the absence of extracellular Ca2+ (0Ca2+) incubation of neurons with thapsigargin eliminated econazole-evoked intracellular Ca2+ increase that was rapidly restored upon incorporating the divalent cation in the external milieu (Fig. 4A). This fast Ca2+ response is consistent with the reported agonistic activity of econazole in store-operated channels (SOC) (28, 29). In the presence of external Ca2+, thapsigargin did not prevent the econazole-evoked cytosolic Ca2+ rise (Fig. 4A). Under these conditions most neurons responded normally to repetitive econazole pulses (Fig. 4A), i.e. increasing the cytosolic Ca2+ level. Nonetheless, we observed a small population of neurons (~25%) where additional pulses of econazole did not produce a Ca2+ response. Presumably, this effect could be mediated by inactivation of SOC or SOC-type response (29). Note that incubation with thapsigargin in the absence of extracellular Ca2+ virtually eliminated the number of neurons responding to econazole (Fig. 4B). Taken together, these results indicate that econazole evokes cytosolic Ca2+ in sensory neurons through a complex mechanism involving regulation of membrane permeability and release from intracellular stores.
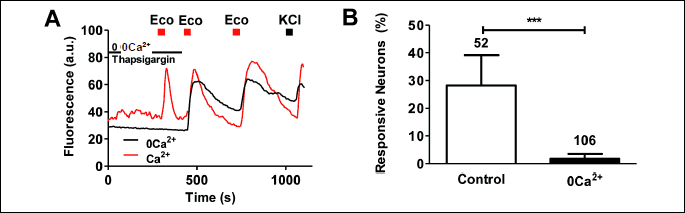
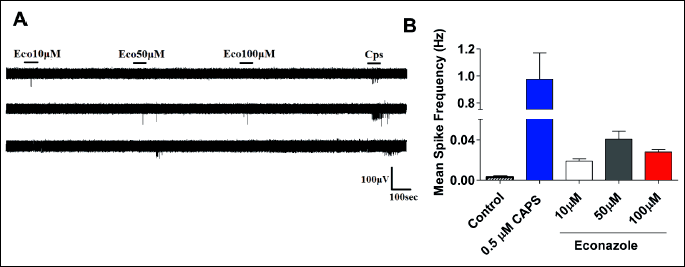
Econazole triggers action potentials in sensory neurons in culture
The econazole-induced rise of cytosolic Ca2+ levels in the sensory neurons implies that the imidazole may modulate neuronal excitability. To investigate the effect of econazole on nociceptor excitability, we evaluated action potential firing using a multi-electrode array (MEA). In control conditions, nociceptors in culture are silent neurons with virtually no firing of action potentials. Electrical activity can be evoked by exposure of these sensory neurons to an excitatory compound such as capsaicin. As seen in Fig. 5, incubation of rat nociceptors with 0.5 µM capsaicin elicited spikes in almost all depicted electrodes (Fig. 5A). This activity can be readily quantified attending to their frequency (Fig. 5B). Akin to the vanilloid, exposure of cultured nociceptors to econazole increased the firing frequency in a dose dependent manner, reaching saturation at 50 µM (Fig. 5B). The imidazole was less potent than capsaicin in activating neuronal activity, although the extent of activation was significant as compared with control conditions. It should be noted that at concentrations higher than 50 µM econazole started to be neurotoxic. Collectively, these results suggest that econazole is able to excite nociceptors, and provide a plausible underlying mechanism for the adverse effects of the treatment in a percentage of patients.
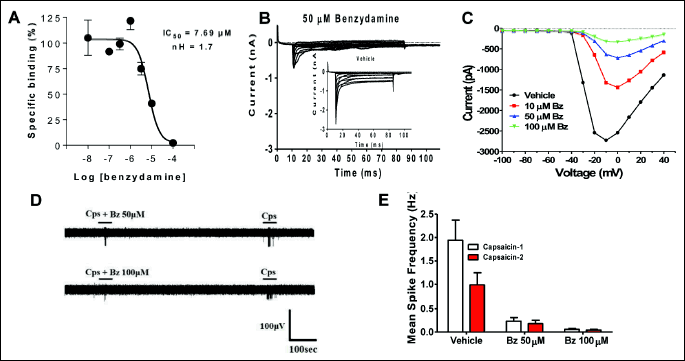
Benzydamine blocks voltage-gated Na+ channels in DRG neurons
Benzydamine is a drug used for attenuating inflammatory skin and mucosae disorders. The mechanism of action has been related to inhibition of inflammatory cytokine production (13, 30-32), although the drug is endowed also with a significant anesthetic effect. To evaluate whether this compound could be used in combination with econazole for reducing the azole-induced neuronal excitability, we first investigated if its anesthetic effect could be related to a block of voltage-gated Na+ currents. As depicted in Fig. 6A, benzydamine is able to bind to site 2 in Na+ channels showing an IC50 of 7 µM. When applied at 50 µM it blocked most of the Na+ inward current elicited by membrane depolarization of nociceptors in culture (Fig. 6B). This blockade activity was concentration dependent showing full inhibition at 100 µM (Fig. 6C). These data demonstrate that benzydamine is a blocker of Nav channels expressed in sensory neurons, and suggest that can be a modulator of nociceptor excitability. Indeed, benzydamine inhibited capsaicin-induced action potentials in sensory neurons (Fig. 6D). The mean spike frequency evoked by capsaicin was virtually abrogated by pre-instillation of benzydamine (Fig. 6E). Thus, benzydamine attenuates nociceptor excitability by preventing the generation and propagation of action potentials through block of Na+ channels.
Benzydamine attenuates econazole evoked excitability in sensitized nociceptors
Pro-inflammatory agents sensitize nociceptors incrementing their sensitivity to excitatory compounds (18, 19), a condition known as neurogenic inflammation (33). This condition may underlie fungal infections, especially of mucosae (34, 35). Thus, we next evaluated whether econazole responses of algesically-sensitized nociceptors were affected by benzydamine. For these experiments, primary nociceptor cultures were sensitized by ATP, a pro-algesic agent that potentiates nociceptor excitability through a P2Y-mediated mechanism that releases Ca2+ from the endoplasmic reticulum (17, 18). As expected, econazole responses were strongly sensitized by ATP as concluded from the comparison of the second and third econazole pulses (Fig. 7A and 7B). Noteworthy, benzydamine did not affect the ATP-induced augment of econazole-evoked Ca2+ responses (Fig. 7B) nor the number of responsive neurons after ATP sensitization (Fig. 7C), consistent with the tenet that the azole-induced Ca2+ responses in sensitized nociceptors were mediated by a channel insensitive to benzydamine. Note that ATP produced a large increase in the Ca2+ that results from the release of Ca2+ from the endoplasmic reticulum.
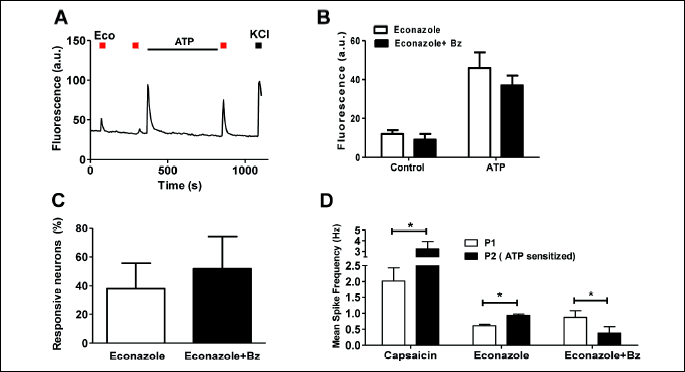
In marked contrast, when the econazole responses were measured using the MEA technology, we observed that the potentiated econazole response upon exposing nociceptors to ATP was significantly attenuated by benzydamine (Fig. 7D), consistent with an inhibitory effect of benzydamine of Nav channels. Thus, under inflammatory conditions econazole appears to evoke stronger neuronal responses, suggesting augmented adverse effects of the drug in sensitized tissue. Blockade of econazole-induced nociceptor excitability with benzydamine should reduce, therefore, the incidence and intensity of burning and itching sensations that, besides being very often associated with the mycosis infection, are exacerbated by the application of the anti-fungic drug econazole.
DISCUSSION
Econazole is an effective anti-mycotic compound widely used for the treatment of cutaneous and vaginal fungal infections. The use of this drug is generally safe, although some complains of burning, itching and irritating adverse effects have been reported by patients, especially in intravaginal treatments. The underlying mechanisms responsible for these side effects have been poorly investigated. Here, we report that econazole, at a clinically relevant concentration (36), directly modulates cytosolic Ca2+ homeostasis in nociceptors as well as their excitability. These effects provide a framework for understanding the reported side effects. An increment of the cytosolic Ca2+ may sensitize a subset of thermal and pruritogenic nociceptors thus leading to burning and itching sensations. In addition, an increased cytosolic Ca2+ may provoke the release of neuronal pro-inflammatory peptides, substance P and calcitonin-gene related peptide, which will provoke neurogenic inflammation, thus directing to erythema and further potentiating the thermal and pruritogenic effects. Overall, the consequence of econazole action on sensory neurons is an increase in their excitability, augmenting the triggering of action potentials in silent nociceptors. Econazole-driven neuronal excitability is more evident in algesically sensitized nociceptors, consistent with the higher Ca2+ fluxes seen in these nociceptors. This result suggests that direct application of the azole compound on sensitized skin or mucosae may exacerbate the sensory side effects. Notably, econazole-evoked nociceptor excitability was significantly reduced by the anesthetic and anti-inflammatory properties of benzydamine, a Nav channel blocker. It should be noted that a similar strategy could be also useful for attenuating the hyperalgesia mediated by other TRP channels (37). In conclusion our observations indicates that a combination of econazole with benzydamine may provide a pharmaceutical formulation for treating cutaneous fungal infections that should exhibit higher tolerability compared to the treatment with econazole alone.
Acknowledgements: Project carried out by means of a grant within the program POR FESR 2007/2013 - avviso pubblico ‘Insieme x Vincere’ (DGR n 580 of 5/12/2012 and subsequent updates: DGR n 103 of 17/05/2013, Det. n G02055 of 12/11/2013 and Det. n G05084 of 17/12/2013), founded by the European Regional Development Fund (ERDF) and Regione Lazio.
Conflict of interest: GM, LP and CM are employees of Angelini S.p.A.
REFERENCES
- Heel RC, Brogden RN, Speight TM, Avery GS. Econazole: a review of its antifungal activity and therapeutic efficacy. Drugs 1978; 16: 177-201.
- Rubio B, Gonzalez R, Karoussos K. Vulvo-vaginal candidosis and its treatment with the new fungicidal substance econazole. J Int Med Res 1980; 8: 436-438.
- Milne LJ. A clinical study of econazole cream in the treatment of fungal skin infections. J R Coll Gen Pract 1982; 32: 360-364.
- Nadalo D, Montoya C, Hunter-Smith D. What is the best way to treat tinea cruris? J Fam Pract 2006; 55: 256-258.
- Shobana CS, Mythili A, Homa M, et al. In vitro susceptibility of filamentous fungi from mycotic keratitis to azole drugs. J Mycol Med 2015; 25: 44-49.
- Ghelardi E, Tavanti A, Lupetti A, et al. Control of Candida albicans murine vaginitis by topical administration of polycarbophil-econazole complex. Antimicrob Agents Chemother 1998; 42: 2434-2436.
- Furneri PM, Corsello S, Masellis G, et al. Econazole-polycarbophil, a new delivery system for topical therapy: microbiological and clinical results on vaginal candidiasis. J Chemother 2008; 20: 336-340.
- Robinson AJ, Wilson JD, Spencer RS, Kinghorn GR. Econazole nitrate (150 mg) single dose vaginal pessary compared with clotrimazole (10%) single dose vaginal cream to treat women with vaginal candidiasis. Genitourin Med 1989; 65: 201-202.
- Elewski BE, Vlahovic TC. Econazole nitrate foam 1% for the treatment of tinea pedis: results from two double-blind, vehicle-controlled, phase 3 clinical trials. J Drugs Dermatol 2014; 13: 803-808.
- Yuca K, Cankaya H, Bayram I, Ozbek H, Kiris M. Local irritant effects of topical oral sprays on oral mucosa in mice. Adv Ther 2006; 23: 98-106.
- Rodriguez-Parkitna J, Korostynski M, Kaminska-Chowaniec D, et al. Comparison of gene expression profiles in neuropathic and inflammatory pain. J Physiol Pharmacol 2006; 57: 401-414.
- Cioli V, Corradino C, Scorza Barcellona P. Review of pharmacological data on benzydamine. Int J Tissue React 1985; 7: 205-213.
- Sironi L, Massimiliano L, Transidico P, et al. Differential effect of benzydamine on pro- versus anti-inflammatory cytokine production: lack of inhibition of interleukin-10 and interleukin-1 receptor antagonists. Int J Clin Lab Res 2000; 30: 17-19.
- Massey T, Derry S, Moore RA, McQuay HJ. Topical NSAIDs for acute pain in adults. Cochrane Database Syst Rev 2010; Jun 16: CD007402. doi: 10.1002/14651858.CD007402.pub2
- Plempel M. Pharmacokinetics of imidazole antimycotics. Postgrad Med J 1979; 55: 662-666.
- Hanel H, Raether W, Dittmar W. Evaluation of fungicidal action in vitro and in a skin model considering the influence of penetration kinetics of various standard antimycotics. Ann NY Acad Sci 1988; 544: 329-337.
- Millikan LE, Galen WK, Gewirtzman GB, et al. Naftifine cream 1% versus econazole cream 1% in the treatment of tinea cruris and tinea corporis. J Am Acad Dermatol 1988; 18: 52-56.
- Camprubi-Robles M, Planells-Cases R, Ferrer-Montiel A. Differential contribution of SNARE-dependent exocytosis to inflammatory potentiation of TRPV1 in nociceptors. FASEB J 2009; 23: 3722-3733.
- Devesa I, Ferrandiz-Huertas C, Mathivanan S, et al. alphaCGRP is essential for algesic exocytotic mobilization of TRPV1 channels in peptidergic nociceptors. Proc Natl Acad Sci USA 2014; 111: 18345-18350.
- Viisanen H, Chapman H, Wei H, et al. Pronociceptive effects induced by cutaneous application of a transient receptor potential ankyrin I (TRPA1) channel agonist methylglyoxal in diabetic animals: comparison with tunicamycin-induced endoplastic reticulum stress. J Physiol Pharmacol 2016; 67: 587-594.
- Jan CR, Ho CM, Wu SN, Tseng CJ. Multiple effects of econazole on calcium signaling: depletion of thapsigargin-sensitive calcium store, activation of extracellular calcium influx, and inhibition of capacitative calcium entry. Biochim Biophys Acta 1999; 1448: 533-542.
- Tunctan B, Altug S, Uludag O, Abacioglu N. Effects of econazole on receptor-operated and depolarization-induced contractions in rat isolated aorta. Life Sci 2000; 67: 2393-2401.
- Kinazaki A, Sakanashi Y, Oyama TM, et al. Micromolar Zn2+ potentiates the cytotoxic action of submicromolar econazole in rat thymocytes: possible disturbance of intracellular Ca2+ and Zn2+ homeostasis. Toxicol in vitro 2009; 23: 610-616.
- Chien JM, Huang CC, Cheng HH, et al. Econazole-evoked Ca2+i rise and non-Ca2+-triggered cell death in rabbit corneal epithelial cells (SIRC). J Recept Signal Transduct Res 2008; 28: 567-579.
- Chang HT, Liu CS, Chou CT, et al. Econazole induces increases in free intracellular Ca2+ concentrations in human osteosarcoma cells. Hum Exp Toxicol 2005; 24: 453-458.
- Tamagnini F, Scullion S, Brown JT, Randall AD. Low concentrations of the solvent dimethyl sulphoxide alter intrinsic excitability properties of cortical and hippocampal pyramidal cells. PLoS ONE 2014; 9: e92557. doi: 10.1371/journal.pone.0092557
- Meseguer V, Karashima Y, Talavera K, et al. Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J Neurosci 2008; 28: 576-586.
- Soboloff J, BergerSA. Sustained ER Ca2+ depletion suppresses protein synthesis and induces activation-enhanced cell death in mast cells. J Biol Chem 2002; 277: 13812-13820.
- Jiang N, Zhang ZM, Liu L, Zhang C, Zhang YL, Zhang ZC. Effects of Ca2+ channel blockers on store-operated Ca2+ channel currents of Kupffer cells after hepatic ischemia/reperfusion injury in rats. World J Gastroenterol 2006; 12: 4694-4698.
- Segre G, Hammarstrom S. Aspects of the mechanisms of action of benzydamine. Int J Tissue React 1985; 7: 187-193.
- Quane PA, Graham GG, Ziegler JB. Pharmacology of benzydamine. Inflammopharmacology 1998; 6: 95-107.
- Modeer T, Yucel-Lindberg T. Benzydamine reduces prostaglandin production in human gingival fibroblasts challenged with interleukin-1 beta or tumor necrosis factor alpha. Acta Odontol Scand 1999; 57: 40-45.
- Planells-Cases R, Garcia-Sanz N, Morenilla-Palao C, Ferrer-Montiel A. Functional aspects and mechanisms of TRPV1 involvement in neurogenic inflammation that leads to thermal hyperalgesia. Pflugers Arch 2005; 451: 151-159.
- Rosen T, Schell BJ, Orengo I. Anti-inflammatory activity of antifungal preparations. Int J Dermatol 1997; 36: 788-792.
- Milani M, Iacobelli P. Vaginal use of Ibuprofen isobutanolammonium (ginenorm): efficacy, tolerability, and pharmacokinetic data: a review of available data. ISRN Obstet Gynecol 2012; 2012: 673131. doi: 10.5402/2012/673131
- Schaefer H, Stuttgen G. Absolute concentrations of an antimycotic agent, Econazole, in the human skin after local application. Arzneimittelforschung 1976; 26: 432-435.
- Wei H, Saarnilehto M, Falck L, et al. Spinal transient receptor potential ankyrin 1 channel induces mechanical hypersensitivity, increases cutaneous blood flow and mediates the pronociceptive action of dynorphin A. J Physiol Pharmacol 2013; 64: 331-340.
A c c e p t e d : December 9, 2016