ALDOSTERONE AND MINERALOCORTICOID RECEPTORS IN REGULATION OF THE CARDIOVASCULAR SYSTEM AND PATHOLOGICAL REMODELLING OF THE HEART AND ARTERIES
Warsaw, Poland
INTRODUCTION
In human beings aldosterone is a principal steroid hormone with mineralocorticoid activity. Aldosterone is produced by aldosterone synthase (CYP11B2), which belongs to the cytochrome P450 (CYP) family and shares 93% homology with its isoenzyme 11-β-hydroxylase (CYP11B1), the enzyme catalysing synthesis of glucocorticoids (1). The main site of aldosterone synthesis is located in the glomerular zone of the adrenal cortex (2). There is also evidence for non-adrenal synthesis of aldosterone, specifically in the brain (3, 4), the heart and the adipose tissue (4-9), although the synthesis of aldosterone in the heart still remains a matter of dispute (10-12). In the adrenal cortex synthesis and secretion of aldosterone are stimulated by potassium ions, angiotensin II (Ang II), angiotensin III (Ang III), vasopressin (AVP), ACTH, β-endorphin, endothelins, adrenomedullin, cholecystokinin and pentagastrin, whereas aldosterone synthesis and secretion may be inhibited by atrial natriuretic peptide, dopamine, somatostatin and nitric oxide (NO) (13-20).
Classical actions of aldosterone are attributed to the kidney and include promotion of transepithelial sodium transport, chloride reabsorption, and potassium and magnesium secretion (21, 22). Thanks to the renal effects, aldosterone plays a fundamental role in maintenance of sodium-potassium balance, and in the regulation of blood volume and blood pressure (22, 23). Dietary salt intake constitutes a key factor determining secretion of aldosterone and potency of its sodium retaining effect (23-25). In rats maintained on normal or high-salt diet plasma aldosterone concentration is low, whereas low-salt diet increases plasma aldosterone concentration and abundance of aldosterone-sensitive sodium channels in the renal tubules (23-25). Moreover, a stimulatory effect of mineralocorticoids on sodium appetite was also demonstrated (26, 27).
Currently, there are solid grounds to believe that aldosterone regulates cardiovascular parameters via its effects exerted in the central nervous system (CNS). Firstly, aldosterone is a steroid that enters the brain in direct proportion to its plasma level (28). Secondly, there is evidence for synthesis of aldosterone within the brain of the rat (4, 29), although it is not yet clear whether it can be synthesized in the normal human brain (30). Finally, mineralocorticoid receptors (MRs) were detected in the regions of the brain that are involved in the cardiovascular regulation (31, 32) and they play an essential role in the central blood pressure regulation (31, 33-37).
In addition, significant number of studies indicate that activation of MRs by aldosterone and glucocorticoids (GCs) directly influences morphology and biochemical composition of vascular and cardiac tissues (38). A large body of evidence shows that disturbances in synthesis and secretion of aldosterone and/or overactivation of MRs play a key role in pathophysiology of cardiovascular diseases (39, 40). Furthermore, recent findings disclosed that aldosterone may also affect the cardiovascular system via MR-independent pathways, which appear to be mediated by G-protein coupled estrogen receptor 30 (GPER, GPR 30) (41-43).
The main purpose of the present review is to summarise current knowledge concerning mechanisms of action of aldosterone in the heart, vessels, and in the brain cardiovascular regions, and to discuss consequences of excessive stimulation of the cardiovascular system by mineralocorticoids. We also discuss positive and negative consequences of application of the treatment interfering with the synthesis or action of aldosterone in the cardiovascular pathology. The detailed analysis of the cellular mechanisms of action of mineralocorticoids has been provided elsewhere in several excellent review articles (31, 39, 41, 44-48).
MINERALOCORTICOID RECEPTORS
Distribution of mineralocorticoid receptors
In the kidney, aldosterone plays its major physiological role via MRs located in the distal portion of the nephron, namely in the distal convoluted tubule, connecting tubule and the cortical and medullary portions of the collecting duct (49). Renal MRs have been detected in the principal cells, intercalated cells, mesangial cells, podocytes, fibroblasts, renal endothelium and vasculature (49-52). Apart from the kidney, MRs are expressed in the vascular endothelium and vascular smooth muscle cells (VSMCs) of several vascular beds including the aorta, coronary vessels, mesenteric and renal interlobar arteries (38, 44, 53-55). In the heart, MRs are also expressed in cardiomyocytes, fibroblasts, and inflammatory cells, such as macrophages and T-lymphocytes (38, 44, 55-59). Furthermore, MRs are also expressed in neurons, microglia and astrocytes of the cerebral cortex, limbic system and cardiovascular regions of the brain – specifically in the brainstem, the hypothalamus and circumventricular organs (60-63).
Genomic actions of aldosterone
The classical genomic MRs belong to transcription factors activated by steroids and are involved in the regulation of gene expression (64). They share structural homology with other intracellular steroid hormone receptors for GCs, progesterone, androgens and estrogens (64-66). Genomic MRs are principal receptors for aldosterone; however, they have low selectivity and except for aldosterone, they bind other mineralocorticoids, such as deoxycorticosterone (DOC) and GCs (38).
The genomic MRs, which are engaged in induction or suppression of the transcription processes, are composed of three domains: the N-terminal transactivation domain (NTD), DNA-binding domain (DBD) located in the central part of the receptor, and the ligand binding domain (LBD) located at the C-terminal part of the receptor (64). Under resting conditions complexes formed with heat shock proteins (HSP 70, HSP 90) keep MRs inactive in the cytoplasm. Binding of the ligand with LBD causes dissociation of HSP to the cytoplasm, and this results in activation of MRs due to a change in conformation of their structure. Next, MRs shift into the nucleus and associate with hormone response element (HRE) or negative steroid response element (nSRE) DNA sequences in the promoter region of the target genes (64, 67). Initiation of transcription and translation processes by aldosterone results in synthesis of so-called aldosterone induced proteins. The process of activation is regulated by a specific combination of coregulatory proteins, such as: coactivators (SRC, PBP/TRAP220, CBP) and corepressors (NCoR, SMRT) (64). As the mechanism of action of the ligands via MR activation has a multistage character, the effects of genomic action of aldosterone can be noted not earlier than after one hour, and they may last for several hours (64, 68).
Non-genomic actions of aldosterone
The non-genomic actions of aldosterone induce rapid cellular responses that are caused by activation of MR-dependent and MR-independent pathways (42, 69). The biological effects of non-genomic actions of aldosterone develop rapidly within seconds to minutes. They appear to play particularly significant role in the brain, especially in the hippocampus and the brainstem, as well as in the cardiomyocytes, endothelial cells and VSMCs (43, 46, 48, 64, 70-73).
The MR-dependent non-genomic actions of aldosterone are mediated by MRs associated with the cellular membrane by scaffolding proteins, such as striatin and caveolins, and transactivation of several G-protein coupled receptors and receptor tyrosine kinases (48), such as insulin-like growth factor 1 receptor (IGF1R), platelet derived growth factor receptor (PDGFR), epidermal growth factor receptor (EPGFR), and angiotensin type 1 receptor (AT1R) (48). It is noteworthy that both MRs and AT1Rs are upregulated in the left ventricle of the infarcted heart (40, 74) and in the CNS of hypertensive and chronically stressed rats (32, 75) as previously reported by us, and that MR-dependent transactivation of AT1Rs was shown to participate in cardiac remodelling and fibrosis (76, 77) as well as in sympathoexcitation (78).
Inhibition of MRs with spironolactone or eplerenone limit the MR-dependent non-genomic effects of aldosterone; however, inhibitors of transcription and translation do not affect the response (42). Stimulation of these receptors accounts for rapid effects engaging numerous cell signalling pathways; however, the molecular mechanism of their activation has not been fully elucidated yet (48, 64). Thus far, it has been shown that rapid effects of aldosterone are associated with activation of protein kinase C (PKC), cyclic adenosine 3’5’-monophosphate (cAMP) and phosphoinositide 3-kinases (PI3K) with further downstream activation of numerous cell-specific kinases, ion channels and pumps (41, 42). Specifically, evidence has been provided for the involvement of inositol triphosphate (IP3), mitogen activated protein kinase (MAPK) phosphatases (MKP-1), cSrc kinase, extracellular signal-regulated kinase (ERK), NADPH oxidase and induction of ROS, sodium hydrogen antiporter (NHE-1), and Na+/K+-ATP-ase (79-85). The biological effects of non-genomic effects induced by aldosterone can last either for a short time, as the ino- and chronotropic effects in the heart or isolated cardiomyocytes (86) and rapid changes in renal handling of sodium ions (87), or chronically persist, as in the process of vascular calcification (88). A growing body of evidence indicates that some of rapid effects of minceralocorticoids in the renal tubules, endothelial cells and the CNS cells involve aldosterone-dependent non-genomic activation of epithelial sodium channels (ENaC), which belong to superfamily of cation channels that can be blocked by amiloride (34, 87, 89, 90).
The MR-independent non-genomic actions of aldosterone are not inhibited by classical MR antagonists. Thus, currently approved pharmacotherapy with spironolactone or eplerenone cannot mitigate detrimental effects of aldosterone mediated by these pathways (42, 69). A growing body of evidence indicates that these non-genomic actions of aldosterone are mediated by activation of GPER, a membrane receptor that is also involved in signalling the non-genomic effects of estrogen. Contrary to the notion of co-activation of GPERs via MR-striatin pathway (48), Ashton et al. provided robust evidence that activation of ERK1/2 and generation of superoxide by cardiomyocytes are exclusively driven by selective activation of GPERs, independently of genomic signaling by MRs (42, 43). The selective activation of GPERs was achieved with pegylated aldosterone, which does not cross cell membrane, thus it interacts only with receptors on the cell surface (42, 43). In addition, the authors showed that GPER-mediated non-genomic effects of aldosterone do not amplify genomic actions of this mineralocorticoid, as expression of aldosterone-regulated genes, Sgk-1 and PAI-1, induced by aldosterone was not affected by GPER antagonist G36 (43). The importance of selective activation of genomic and non-genomic responses triggered by aldosterone under ex vivo conditions was supported by findings that selective activation of GPERs and subsequent ROS generation is ineffective in augmenting post-ischemia myocardial injury, suggesting a critical role of intracellular MR activation in development of acute ischemia-reperfusion cardiac injury (43). Thus, it is suggested that activation of intracellular MRs is required for manifestation of detrimental effects of aldosterone in the ischemic cardiac tissue (43). Furthermore, selective activation of GPERs with pegylated aldosterone was shown to disassociate MR-striatin complexes without nuclear translocation of MRs, whereas non-selective activation of MR with aldosterone resulted in ablation of MR-striatin complexes and a shift of MRs into the cell nucleus (43). These findings suggests that GPERs presumably participate in preparing MRs for binding with its agonists (43). In addition to the effects of GPER in cardiomyocytes and ischemic myocardium, stimulation of GPERs by aldosterone was shown to induce VSMC contractions as well as apoptosis of VSMCs and endothelial cells (42, 48, 91). It has also been shown that aldosterone-mediated activation of GPERs in the nucleus ambiguus increases neuronal activity and tone of the cardiac vagus (72) as well as activates the NTS neurons with subsequent increase in sodium intake (73) in MR-independent manner. In this light, the MR-independent effects of aldosterone warrant further studies, which will determine therapeutic potential of GPERs and modulation of genomic effects of aldosterone.
The role of non-genomic actions of aldosterone in the cardiovascular system has been intensively investigated in recent years. This research has been summarized by several timely reviews (69, 92-94). Specifically, interactions of non-genomic and genomic effects of aldosterone in different organs and tissues and putative significance of these actions in cardiovascular diseases has been discussed in the review paper of Mihailidou et al. (42).
11β-HYDROXYSTEROID DEHYDROGENASE
Properties of 11β-HSD types 1 and 2
Aldosterone and cortisol bind with MRs with similar affinity (95-99), whereas concentration of GCs in plasma is 100 (free) to 1000 (total) times higher than that of mineralocorticoids (64). Thus, in most of the tissues, probability of binding of GCs to MRs and GC receptors (GRs) is significantly greater than the chance of binding of aldosterone. Cortisol in humans and corticosterone in rodents contain a hydroxyl group at carbon C-11 and are active steroid hormones. Conversion of this group into a keto group results in the transformation of active steroids into inactive cortisone and 11-dehydrocorticosterone, respectively (100). The conversion can be catalysed by two microsomal isozymes, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) (99). The former compound is a bidirectional enzyme with dehydrogenase activity that catalyses conversion of GCs into the inactive ketoanalogues as well as reductase activity that converts the inactive metabolites to active GCs (101-104). In contrast, 11β-HSD2 manifests exclusively unidirectional dehydrogenase activity and catalyses oxidation of cortisol into inactive cortisone. At low concentrations or in the absence of 11β-HSD2, MRs interact with cortisol, which is present in significantly higher concentration in plasma and tissues than aldosterone. Thus, a tissue-specific expression of these two isoforms of 11β-HSD determines the pre-receptor regulation of GR and MR activation and tissue-specific actions of aldosterone (102, 103, 105). The direction of enzymatic activity of 11β-HSD1 also depends on availability of cofactors, in particular oxidised/reduced forms of NADP+/NADPH. 11β-HSD1 has reductase activity in the presence of NADPH as a cofactor, whereas it shows dehydrogenase activity when NADP+ is available (106). The production of NADPH occurs in the endoplasmic reticulum lumen and depends on the activity of other enzymes. For instance, it was shown that its production requires presence of hexose-6-phosphate dehydrogenase (H6PDH), which causes fivefold increase in stimulation of oxidoreductase activity and sixfold decrease in dehydrogenase activity of 11β-HSD1 (107). When H6PDH is absent, 11β-HSD1 acts mainly as a dehydrogenase. Nonetheless, in vivo 11β-HSD1 has mostly reductase activity, thus it facilitates activation GRs (108-110).
Importance of tissue-specific expression of 11β-HSD2 for activation of mineralocorticoid receptors
The affinity of 11β-HSD2 to substrates is approximately 100-times greater than the one of 11β-HSD1 (99, 111). Consequently, relatively small amount of 11β-HSD2 is sufficient for protection of MRs from binding with GCs (99, 109). Thus, when both isoforms of 11β-HSD are expressed, the higher binding affinity of 11β-HSD2 to the substrates and its specific intracellular localisation cause that 11β-HSD2 plays a dominant role in metabolism of corticosteroids (105, 109). Thanks to availability of 11β-HSD2 in epithelial cells of the distal portion of the nephron and in the VSMCs, these cells respond specifically to mineralocorticoids in spite of the presence of GCs (38, 112-115). On the other hand, the lack or low expression of 11β-HSD2 and presence of 11β-HSD1 in the inflammatory cells and adipocytes make MRs in these cells chief targets for highly abundant cortisol in humans and corticosterone in rodents (38, 45).
In the heart the expression of 11β-HSD2 is relatively low and MRs in the cardiomyocytes are occupied mainly by endogenous GCs (112, 116, 117). Available data suggests that GCs may act as antagonists for cardiomyocyte MRs, thus they may confer some protective effects under condition of high aldosterone levels (118-121). This notion is further substantiated by preclinical studies indicating that there is an association between 11β-HSD2 activity and the cardiovascular pathology. Specifically, selective overexpression of 11β-HSD2 in cardiomyocytes, which leads to selective activation of MRs by aldosterone, resulted in cardiac hypertrophy, fibrosis, heart failure and early death suggesting that endogenous GCs occupying MRs provide protective effects against aldosterone-mediated activation of MRs in the cardiac muscle (122). Nonetheless, under conditions of increased oxidative stress, such as in the heart failure or hypertension, GC-MR complexes may become activated by reactive oxygen species (ROS) (39). In line with this, it was shown that activation of MRs by GCs results in coronary vessels and heart muscle injury in a low-aldosterone model of hypertension and heart failure, which in turn is in contradiction with protective action of GCs in the cardiac muscle (123).
Moreover, there is a relationship between the decreased activity of 11β-HSD2 and hypertension in humans (124, 125). DNA methylation as well as epigenetic suppression of 11β-HSD2 cause the increase in the ratio of active 11β-HSD1 to 11β-HSD2, thereby escalating action of GCs on the MRs and GRs and promoting development of the cardiometabolic syndrome. Therefore, it is suggested that 11β-HSD1 constitutes the pharmacological target for the development of selective antagonists that presumably should alleviate pathological consequences of diseases associated with overstimulation of GRs, such as obesity, diabetes type 2 and cardiometabolic syndrome (39, 64, 126).
In the brain, a relatively high expression of 11β-HSD2 was found in the nucleus of the solitary tract (NTS) (36, 64). Some reports show that 11β-HSD2 mRNA is also expressed in the hypothalamic paraventricular nucleus (PVN), the supraoptic nucleus (SON) and the subfornical organ (SFO) (127, 128). Thus, MRs in the key structures of the CNS involved in the cardiovascular regulation may be selectively targeted by aldosterone. However, expression of 11β-HSD1 dominates in other regions of the brain (36, 64), where GCs seem to play the dominant role in MR activation.
ALDOSTERONE AND THE HEART
Effects of cardiac mineralocorticoid receptors overstimulation
Expression of MRs was detected in human cardiac tissues obtained from the atria and ventricles, and it is reasonable to assume that these receptors are stimulated by aldosterone circulating in the blood (40, 64, 129). Besides, de novo synthesis of aldosterone by CYP11B2, aldosterone synthase, was detected in the atria and in ventricular cardiomyocytes, thus it is suggested that locally synthesised aldosterone may play a role in the pathogenesis of cardiac hypertrophy and fibrosis (5, 64, 67). Nonetheless, expression of the aldosterone synthase in the cardiac muscle is several orders of magnitude lower than in the adrenals, suggesting that it is mainly the blood-borne aldosterone that binds to MRs in the heart (67).
Based on results from experiments in mice with cardiomyocyte specific overexpression of 11β-HSD2, it was postulated that under pathological conditions excessive binding of mineralocorticoids with MRs leads to cardiac injury (122). Moreover, in a study by Silvestre et al. (130) investigating interactions between aldosterone, MRs and AT1Rs, it was shown that in rats with myocardial infarction (MI) the expression of mRNA for aldosterone synthase increases two-folds and aldosterone level almost quadruples in the residual intact myocardium. In addition, the upregulation of aldosterone synthase was dependent on activation of AT1Rs, whereas blockade of MRs significantly limited the post-infarct deposition of collagen in the heart (130). The results from animal studies were confirmed by findings in patients with congestive heart failure, which proved that increased expression of MR mRNA and protein was present in cardiomyocytes obtained from the left ventricle of the failing heart (131). Increased expression of MRs was also reported in the atrial tissue obtained from patients with atrial fibrillation (AF) (129). Furthermore, both in AF patients and in a mouse model of AF increased expression of 11β-HSD2 and aldosterone-dependent increase in connective tissue growth factor (CTGF) were observed (132).
A growing body of evidence indicates that under pathological conditions proinflammatory and fibrogenic phenotypes develop in the heart and coronary vessels (41, 133-135). In a hypertensive model of uninephrectomized rats exposed to chronic treatment with aldosterone and salt, aldosterone-dependent remodelling and fibrosis of the heart and coronary arteries was mediated by activation of NADPH oxidase, production of ROS and induction of the proinflammatory response manifested by cardiac expression of proinflammatory mediators and recruitment and accumulation of macrophages in the vessels and cardiac tissue (133, 134). In addition, it was shown in a genetic mouse model of cardiac-specific hyperaldosteronism and systemic hypertension that fibrotic changes in the heart involve increased expression of chemoattractant proteins (galectin 3, monocyte chemoattractant protein 1, osteopontin), leading to infiltration of the cardiac tissue with macrophages (136). Furthermore, cardiac fibrosis at least partially depends on aldosterone-induced decrease in antifibrotic factors, such as brain natriuretic peptide (BNP) and bone morphogenic peptide (133, 134, 136). Additionally, aldosterone contributes to progression of cardiac fibrosis by increase in expression of PAI-1 and tissue inhibitor of metalloproteinase 1 (TIMP-1), which are dependent on aldosterone-mediated suppression of natriuretic peptides (67, 136).
Together with inflammatory dependent fibrogenic response, aldosterone promotes fibroblasts proliferation and hypertrophy as well as collagen and fibronectin synthesis due to activation of several essential enzymatic pathways. Among these actions are: phosphorylation of ERK and activation of ERK/MAPK and RAS-Raf-MEK-ERK signalling cascades, activation of p38 mitogen-activated protein kinase (p38MAPK), enhancement of expression of transforming growth factor-β (TGF-β) and suppression of expression of inducible nitric oxide synthase (iNOS) (67, 137, 138). Experiments in a mouse model of cardiomyocyte-specific overexpression of human MRs revealed that prolonged overstimulation of cardiac MRs leads to endothelial dysfunction of coronary vessels manifested by decreased sensitivity to NO-mediated vasodilatory responses to acetylcholine. This desensitization was associated with increased cardiac levels of ROS, cardiac NADPH oxidase (NOX) activity, and increased expression of the NOX subunit gp91phox (139). The negative effects of MR overstimulation could be prevented by treatment with MR antagonist, antioxidant vitamins E and C, or NADPH oxidase inhibitor. Together, these findings suggest that overstimulation of MRs in cardiomyocytes may activate a paracrine mechanisms, which lead to NOX-dependent increase in ROS in the coronary vessels (139).
Increase in fibronectin and cardiac fibroblast-mediated collagen synthesis as well as decrease in matrix metalloproteinase (MMP) activity are further enhanced by local intracardiac interactions between aldosterone and Ang II, as aldosterone upregulates AT1R expression resulting in activation of NADPH oxidase and ROS generation (138, 140-142). This aldosterone-mediated progression of cardiac pathological remodelling leads to decreased cardiac compliance, diastolic and systolic dysfunction, propensity for arrhythmias and may result in sudden cardiac death (138, 143).
Furthermore, conditional overexpression of MRs in cardiomyocytes in mice results in cardiac infiltration of inflammatory cells, cardiac fibrosis, and prolongation of action potential duration due to downregulation of the transient outward potassium current (Ito) with simultaneous elevation of L-type calcium channel activity. These changes lead to prolongation of PQ interval and QRS complex, ventricular arrhythmias and greater mortality rate (144). In this line, experiments on isolated cardiomyocytes revealed that aldosterone exerts positive chronotropic effect (145). It was also shown that overexpression of MRs in the cardiomyocytes leads to increased beating frequency (146), which may contribute to proarrhythmic phenotype.
Effects of selective suppression of cardiac mineralocorticoid receptors
Experiments with cardiomyocyte-specific down-regulation or deletion of MRs highlight the importance of excessive stimulation of MRs in the cardiac pathophysiology. It has been shown that deletion or inactivation of the MR gene lessened progression of left ventricular dilatation, as well as reduced the development of myocardial hypertrophy and heart failure in animal models of MI and pressure overload-induced cardiac hypertrophy (57, 144, 147-149). Furthermore, Fraccarollo et al. (57) showed that genetic ablation of MRs in the cardiomyocytes prevented post-infarct generation of myocardial and mitochondrial superoxide and abolished upregulation of the NADPH oxidase subunits (Nox2 and Nox4), improved healing of the MI, increased capillary density of the non-infarcted myocardium and thickness of the infarction zone scar. Thus, the cardiomyocyte-specific MRs ablation prevented adverse cardiac remodelling associated with cardiac hypertrophy and fibrosis (57). These changes were accompanied by improvement in hemodynamic parameters, such as left ventricular filling pressure, left ventricular ejection fraction, end systolic and end diastolic left ventricular volumes as well as by reduction in pulmonary oedema (57).
The crucial role of MRs in cardiac remodelling and fibrosis was also shown in 11-deoxycorticosterone (DOC)/salt induced hypertension in a model of MR-null mice, not expressing MRs in the myocardium. In comparison to wild type mice, eight-week exposure of mice lacking cardiac MRs to DOC/salt treatment resulted in lower expression of profibrotic factors (PAI-1, VEGFa, TGF-β, and integrin β1), inflammatory markers (MCP-1, CCR5, CD14, and CD81), and oxidative stress markers (p22phox) with eventual reduction of cardiac fibrosis (38, 150).
Role of mineralocorticoid receptors in cardiac remodelling in hypertension
Experimental studies strongly support involvement of MRs in cardiac remodelling in aldosterone and Ang II dependent hypertension, and in essential hypertension. Specifically, rats on high salt intake treated with aldosterone infusion show increased expression of chief prooxidant and proinflammatory-related genes, such as NFκB, p38MAPK, and TGF-β1, as well as elevated ROS generation in cardiomyocytes and peripheral blood mononuclear cells (PBMCs) that eventually lead to reparative cardiac fibrosis and coronary vasculopathy (151, 152). Accordingly, it was suggested that analysis of biochemical profile of PBMCs may have a predictive value for evaluation of cardiovascular risk prior to evident clinical symptoms (151). Furthermore, blockade of MRs with eplerenone significantly reduced oxidative stress in the heart and kidney of mice rendered hypertensive by chronic infusion of Ang II (153).
The study of Konishi et al. (154) showed significantly higher expression of 11β-HSD2, MRs and collagen 1 and collagen 3 in the left ventricle of stroke-prone spontaneously hypertensive rats (SHR-SP) with malignant hypertension, suggesting that aldosterone and MRs may play a role in remodelling of the heart in essential hypertension.
ALDOSTERONE AND VESSELS
Effects of mineralocorticoid receptors’ stimulation in vascular wall
Vascular MRs are effectively and selectively stimulated by aldosterone. In vessels aldosterone exerts both rapid non-genomic, and delayed genomic effects, and participates in the regulation of the vascular tone and remodelling of the vascular wall, as manifested by VSMCs’ proliferation and hypertrophy and vascular fibrosis (155, 156).
Binding of aldosterone to MRs in the arterial wall leads to increased expression of MMPs, TGF-β1, CTGF and galectin-3 with eventual remodelling of the extracellular matrix (ECM), manifested by increase in collagen to elastin ratio as well as increase in content of fibronectin and proteoglycans. These aldosterone-induced changes translate into impaired elasticity and compliance resulting in the arterial stiffness (157). Evidence from experiments in mice with conditional inactivation of MRs in VSMCs indicates that the remodelling of the arteries and development of hypertension is mostly dependent on activation of MRs in the VSMCs (158). In contrast to MRs expressed in the VSMCs, it seems that endothelial MRs are not significantly involved in the regulation of the arterial blood pressure and development of hypertension (38, 52, 159).
Numerous studies indicate that aldosterone promotes proliferation of VSMCs through activation of kinases – the classical MAPK-ERK1/2 and Big MAP kinase 1 (BMK1) pathways (160, 161). Furthermore, in human VSMCs and in the aorta of rats aldosterone significantly increases expression of MDM2, one of the oncoproteins with anti-apoptotic properties that is involved in the hypertrophy and hyperplasia of VSMCs (162-164). Similarly, aldosterone-dependent increase in galectin-3 expression leads to cell proliferation, adhesion, and fibrosis with subsequent pathological remodelling of the arterial wall. Recent studies showed that overexpression of galectin-3 promotes aldosterone-induced collagen 1 synthesis, whereas galectin-3 inhibition attenuates this process (157). Thus, galectin-3 is required for inflammatory and fibrotic responses to aldosterone in VSMCs and constitutes an important biomarker of cardiovascular fibrosis (157, 165). Furthermore, chronic blockade of MRs with eplerenone, selective MR antagonist, protects vascular wall from aldosterone-induced pathological remodelling, as it was shown in the rat model of adrenal aldosterone-producing adenoma (163). In addition, emerging evidence indicates that aldosterone-induced remodelling of the arterial wall is in part dependent on activation of placental growth factor (PGF) – FMS-like tyrosine kinase 1 (Flt1) pathway (166).
Under physiological conditions, VSMC MRs contribute to the regulation of vascular tone in large arteries and participate in maintenance of normal blood pressure. A recent study in mice with conditional inactivation of MRs in the VSMCs showed that MRs expressed in VSMCs play essential role in determining magnitude of contractions and relaxations of aorta induced by extracellular calcium and NO, respectively. Their presence is also necessary for efficient expression of VSMC contractile proteins and their regulators (53). Furthermore, aldosterone-dependent activation of Nox1 subunit of NADPH oxidase and formation of ROS is responsible for hypercontractility of the resistance arteries obtained from stroke-prone spontaneously hypertensive rats (167). By activating MR-dependent ROS formation, aldosterone participates in the development of inflammatory response in the vascular wall, eventually leading to vascular injury (167). Furthermore, aldosterone may stimulate thrombogenesis via activation of platelets and by reducing fibrinolysis, whereas the treatment with MR antagonists hinders these effects (168-173).
Interaction of mineralocorticoid receptors and AT1Rs in the vascular wall
Ang II is the most significant effector of the RAS (174). It exerts its physiological actions through angiotensin type 1 (AT1R) and type 2 (AT2R) receptors, which belong to membrane-bound G-protein coupled receptor family (GPCR) (175). Numerous studies revealed that Ang II is critically involved in remodelling of the cardiovascular system. in vitro studies showed that Ang II stimulates the production of ECM components and promotes proliferation of VSMCs (175, 176). Furthermore, evidence from in vivo experiments showed that Ang II-induced vascular fibrosis and remodelling are mediated by AT1R and involve AT1R-dependent increase in expression of collagen, fibronectin, osteopontin and proteoglycans. In addition, activation of AT1Rs in the coronary arteries promotes development and progression of atherosclerosis, eventually leading to MI, heart failure and stroke (175, 177, 178). A large body of evidence also indicates that chronic overactivation of vascular AT1R results in hypertension (175, 179, 180).
There is a close relationship between Ang II signalling pathways and activation of MRs in the vascular wall. Thus, hypertension occurring in response to administration of Ang II at least partially depends on activation of MRs in the VSMCs (38).
Moreover, it has been shown that in hypertensive transgenic Ren-2 rats overexpressing the mouse renin gene and high concentration of plasma Ang II, mitochondrial abnormalities, enhanced apoptosis, increased intracellular lipid accumulation and vascular injury are largely mediated by increased activation of MRs (160). This is manifested by significant attenuation of elevated NADPH oxidase activity, lipid peroxidation, and apoptosis as well as by reduced expression of AT1R and renin in vascular wall after inhibition of MRs with spironolactone in this model of hypertension (160).
Emerging evidence indicates that MR upregulation may also cause pathological alterations in venous vessels. For instance, remodelling of venous grafts after implantation into the arterial circulation is dependent on RAS and MRs. Specifically, elevated expressions of mRNAs encoding the MRs, 11-β-HSD2, AT1R, and the angiotensin-converting enzyme (ACE) were demonstrated in human venous VSMCs obtained from the saphenous veins used for coronary artery by-pass surgery (181). Furthermore, in a mouse model of the inferior vena cava implants into the abdominal aorta, it was shown that expression of MRs is upregulated after vein grafting. Moreover, blockade of MRs significantly decreased remodelling of the venous wall, which was manifested by decrease in graft intima-media thickness, reduced fibrosis, and lower infiltration of inflammatory cells in the wall of the venous graft (181), suggesting a critical role of MRs in the vascular remodelling of the venous grafts.
CENTRALLY MEDIATED CARDIOVASCULAR EFFECTS OF ALDOSTERONE
Aldosterone and mineralocorticoid receptors in the central nervous system
Multiple studies argue for significant role of aldosterone in the central regulation of the cardiovascular system, however thus far there is no full consensus about the origin of aldosterone acting on the central cardiovascular neurons. Available evidence indicates that aldosterone may be locally synthesised in the brain. Specifically, expression of the mRNA for the aldosterone synthase was demonstrated in the hypothalamus, hippocampus, amygdala, cerebrum, and cerebellum (3, 28, 182). Nevertheless, the hormone may also have an access to the brain via the circumventricular organs, and to some extent it can penetrate the blood-brain barrier (36, 183).
Findings of a recent study by Wang et al. (127) indicate that blood-derived aldosterone may influence local production of aldosterone in the hypothalamic cardiovascular nuclei, namely PVN and SON, via MR- and AT1R-dependent mechanisms operating in the subfornical organ (SFO), and that aldosterone upregulates AT1R signalling in the PVN through activation of MRs. Furthermore, stimulation of the SFO and its downstream connections to the hypothalamic PVN neurons by circulating aldosterone appears to be necessary for development of hypertension in rats maintained on increased salt intake and chronically infused with aldosterone (127).
Expression of MRs is high in several regions of the brain participating in the regulation of blood pressure (31, 36, 64). Especially noteworthy is presence of MRs in the sympathetic preautonomic neurons of the PVN (62) as these neurons send projections to several autonomic motor nuclei in the brainstem and the spinal cord (184, 185). In addition, MRs are expressed in the SFO, one of the circumventricular organs lacking the blood-brain barrier with extensive connections with the PVN. Activation of MRs in the SFO by aldosterone present in the bloodstream leads to excitation of preautonomic neurons present in the hypothalamus (127). Furthermore, there is a large body of evidence indicating that MRs expressed in the NTS participate in the elevation of salt appetite induced by aldosterone (186, 187). In addition, MR expressing neurons in the NTS project to the limbic-forebrain circuits and affect arousal and motivational behaviour (187). MRs expressed in the limbic system and the forebrain affect memory processes, appraisal and coping strategies under stress conditions; however, it seems that these MRs are mainly targeted by GCs (187).
Cardiovascular effects of stimulation of mineralocorticoid receptors and AT1Rs in the brain
Functional studies show that stimulation of the central MRs play a significant role in endocrine responses to stress and in regulation of mood and cognition (31, 64, 187). Stimulation of MRs in the brain also activates sympathetic nervous system and increases arterial blood pressure, and these effects are associated with stimulation of the brain RAS and the induction of oxidative stress as well as activation of aldosterone-MR-ENaC-ouabain cascade in the CNS (78, 183, 188-191).
Furthermore, it was shown that blood-borne aldosterone increases sympathetic activity via MR-dependent activation of p44/42 MAPK signalling pathway in the PVN neurons, which are crucial for aldosterone-induced sympathetic response. These aldosterone-induced MAPK activation and sympathetic excitation partially depend on AT1R stimulation in the brain (78).
Taken together, aldosterone, locally synthesised or blood-borne, and brain MRs contribute to sympatho-excitation. This chronically increased sympathetic activity promotes development and progression of hypertension and is associated with the end-organ damage as well as cardiovascular complications such as MI, stroke and heart failure (192, 193). Similarly to aldosterone, i.e. via MAPK induction, Ang II increases AT1R expression in the PVN, contributing thereby to sympathetic excitation in heart failure (78).
In the brain, the pre-receptor control of GCs binding to MRs by 11β-HSD2 cannot be efficient due to relatively poor expression of this enzyme in the CNS. It is suggested that in most instances brain MRs are occupied by GCs. However, higher expression of 11β-HSD2 in the NTS, PVN, SON and SFO suggests that these cardiovascular centres may be selectively targeted by aldosterone (36, 64, 127, 128). In addition, aldosterone may become a dominant ligand interacting with MRs in pathological conditions, in which its concentration increases excessively or its action is facilitated by concomitant factors, such as sodium overload, Ang II, oxidative stress or cytokines (39, 64). Main steps of synthesis and metabolism of aldosterone and its interaction with angiotensins and other cardiovascular factors are illustrated in Figs. 1 and 2.
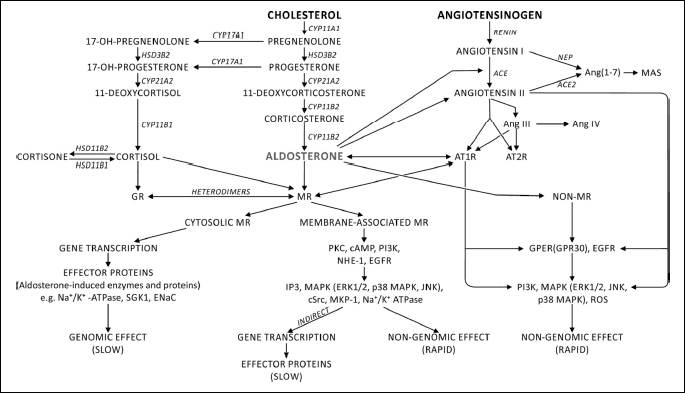
The arrows indicate the direction of interactions and their stimulating character. Abbreviations: CYP11A1, cytochrome P450 family 11 subfamily A member 1; CYP17A1, cytochrome P450 family 17 subfamily A member 1; CYP21A2, cytochrome P450 family 21 subfamily A member 2; CYP11B1, cytochrome P450 family 11 subfamily B member 1; CYP11B2, cytochrome P450 family 11 subfamily B member 2; HSD11B1, hydroxysteroid 11-beta dehydrogenase 1; HSD11B2, hydroxysteroid 11-beta dehydrogenase 2; HSD3B2, hydroxysteroid 3-beta dehydrogenase 2; ACE, angiotensin converting enzyme; MR, mineralocorticoid receptor; GR, glucocorticoid receptor; AT1R, angiotensin type 1 receptor; AT2R, angiotensin type 2 receptor; MAS, proto-oncogene G protein-coupled receptor; ENaC, epithelial sodium channel; SGK1, serum and glucocorticoid regulated kinase 1; PKC, protein kinase C; cAMP, cyclic adenosine 3’5’-monophosphate; PI3K, phosphoinositide 3-kinases; EGFR, epidermal growth factor receptor; NHE-1, N+/H+ exchanger; GPER, G protein-coupled estrogen receptor; GPR30, G protein-coupled receptor 30; IP3, inositol 1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; JNK, c-Jun N-terminal kinase; p38MAPK, p38 mitogen-activated protein kinase; cSrc, cytoplasmic tyrosine-protein kinase; MKP-1, mitogen-activated protein kinase phosphatase 1.
ALDOSTERONE IN CARDIOVASCULAR PATHOLOGY
Numerous studies, performed on experimental animals and on patients suffering from cardiovascular pathology, argue for multifarious involvement of aldosterone-MR axis in pathogenesis of cardiovascular diseases. In patients with heart failure concentration of aldosterone in plasma correlates positively with hypertrophy of the left ventricle, and with mortality (194, 195). Furthermore, in hypertensive patients plasma aldosterone level is correlated with vascular stiffness (196). In addition, several studies in rat models and human subjects reveal that in congestive heart failure, and in hypertension without systolic dysfunction of the left ventricle, elevated levels of plasma aldosterone are associated with local production of aldosterone in the heart muscle (7, 130, 197). Both in hypertension and in heart failure, elevated concentration of aldosterone is associated with increased activation of RAS, and several studies have shown that Ang II and stimulation of AT1Rs play essential role in negative effects of aldosterone on the cardiovascular system (28, 120, 130, 182, 198, 199). However, there is also indirect evidence showing that aldosterone may promote hypertrophic remodelling of the heart independently of RAS. Such a possibility is strongly supported by the study performed on hypertensive patients with primary aldosteronism, whose blood pressure was matched with blood pressure of patients with primary hypertension. The patients with primary aldosteronism manifested significantly higher left ventricular wall thickness and left ventricular mass index, as well as longer PQ interval and greater left ventricular concentric remodelling than the patients with essential hypertension despite of significantly lower plasma renin activity (182). Currently, the role of inflammatory processes as a key factor in aldosterone-induced cardiovascular pathology is intensely explored (38, 45, 150, 151, 181, 200).
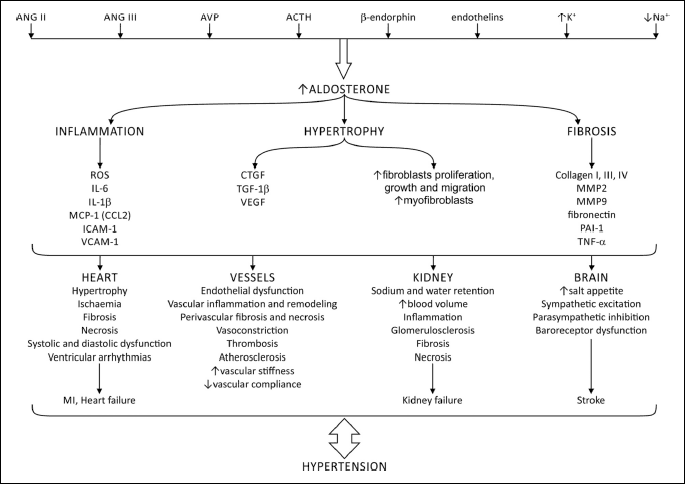
Experimental studies provide evidence that overstimulation of the brain MRs plays a significant role in development of hypertension and heart failure (183, 201). It has been shown that circulating Ang II increases expression of MRs and AT1Rs in the SFO and that this leads to increased production of aldosterone in the hypothalamus, as well as to an enhanced stimulation of AT1Rs in the PVN. Finally, activation of the PVN neurons results in overstimulation of the preautonomic neurons of the sympathetic system innervating the cardiovascular system and in progression of hypertension. In support of this assumption is the finding that elimination of MR or AT1R gene expression in the PVN prevents development of hypertension induced by subcutaneous infusion of Ang II (127, 202). Similar effects were obtained in rats treated with intrabrain infusion of Ang II (203), which suggests a critical MR-dependent central component of Ang II-induced hypertension. In line with these findings in hypertensive animal, there is also evidence that centrally acting aldosterone increases activity of the hypothalamic RAS and participates in generation of increased sympathetic activity in the heart failure (28, 201, 204).
THERAPEUTIC PERSPECTIVES OF INHIBITION OF MINERALOCORTICOID RECEPTORS
The above survey clearly indicates that excessive stimulation of mineralocorticoid receptors plays a critical role in the development of hypertension and cardiac failure. In humans, the pathogenic role of aldosterone was particularly well documented in salt-dependent hypertension, heart failure, and cardiometabolic syndrome (9, 205-208). Several clinical trials were carried out in order to evaluate the effectiveness of inhibition of MRs in cardiovascular patients. In majority of them, classical steroidal MR antagonists spironolactone and eplerenone were used, though there were also attempts to evaluate effectiveness of non-steroidal MR antagonist (49, 172, 205, 209-211).
Several large clinical trials provided a robust evidence for the efficacy of MR antagonists in chronic heart failure patients in NYHA functional class III or IV. Specifically, in the RALES study, blockade of MRs by spironolactone reduced the risk of sudden death from cardiac incidents and from progressive heart failure by 30% (205, 212). Similar results were obtained for eplerenone in the EPHESUS study in patients with chronic heart failure (213). Moreover, in the EMPHASIS-HF study, the benefit of eplerenone treatment in addition to optimal pharmacotherapy with ACE inhibitors, angiotensin-receptor blockers (ARBs), and β-blockers was also confirmed in patients with moderate systolic heart failure and NYHA functional class II symptoms (214).
The efficacy of MR antagonists in patients with primary hypertension is less evident. The recent systemic review of placebo-controlled clinical trials by Tam et al. (215) showed that application of eplerenone as the antihypertensive treatment in patients with primary hypertension results in a modest reduction of systolic and diastolic blood pressure, and the effects of eplerenone on mortality and morbidity are not well supported (215). However, addition of low-dose eplerenone to RAS inhibitors resulted in renoprotective effects in hypertensive patients with non-diabetic chronic kidney disease (CKD) in the EVALUATE Study Group (216). Furthermore, a growing body of evidence indicates that MR antagonists, especially low-dose spironolactone, improve blood pressure control in patients with resistant hypertension (217).
A growing body of evidence indicates that MR antagonists have antiarrhythmic properties in patients with atrial fibrillation (AF) (218). Namely, in the SPIR-AF study, addition of spironolactone to the pharmacotherapy decreased incidence of AF (219). Furthermore, a recent meta-analysis by Neefs et al. suggests that MR antagonists decrease the incidence of new-onset AF and recurrent AF (220).
It should be emphasized that MR antagonists exert also several side-effects, which may hamper their efficacy in cardiovascular patients. The side-effects inherent to administration of spironolactone or eplerenone result mainly from suppression of the renal tubule MRs and secondary hyperkalaemia and renal insufficiency. Usually, the side effects of MR antagonists related to hyperkalaemia do not exceed 10 – 12% of patients (44, 221). Nevertheless, they constitute an important factor limiting the dosage of both these antagonists and impose necessity of frequent laboratory monitoring of potassium and creatinine levels. The risk of hyperkalaemia can be reduced by avoidance of potassium-retaining drugs or potassium-containing foods. Recently, positive effects of administration of potassium-binding substances, such as RLY5016, sodium zirconium cyclosilicate or Patiromer, on plasma potassium level were reported in patients with chronic heart failure and in those with CKD treated with RAAS inhibitors (222, 223). In addition, structural similarity of spironolactone to progesterone and its relatively high affinity for androgen receptors result in progestational and anti-androgenic effects, which are manifested by gynecomastia, breast tenderness, impotence, loss of libido, and irregular menstrual bleedings (224).
Since there are currently only two registered substances, which block MRs, spironolactone and eplerenone, there is a great interest in novel drugs that decrease activation of MRs in patients with cardiovascular diseases. Thus, safety and tolerability of new non-steroidal MR antagonists, in particular apararenone, esaxerenone and finerenone, have been tested in healthy volunteers and patients with heart failure, CKD, diabetes and hypertension. Of these three compounds, finerenone appears to be the most promising non-steroidal MR antagonist (49, 225, 226).
Effectiveness of finerenone in lowering oxidative stress biomarkers in blood was equal to that of spironolactone, whereas a risk of hyperkalaemia and renal dysfunction was lower in patients receiving finerenone. It is believed that finerenone has higher affinity for cardiovascular tissue than for renal tissue (cardiac-to-renal activity ratio) in comparison to spironolactone, however, the hypotensive effectiveness of finerenone is lower in comparison to spironolactone (227, 228). Furthermore, finerenone, was evaluated in the ARTS-HF study in a group of patients with heart failure and reduced LVEF, and with mild or moderate CKD or T2DM (229). Currently, this non-steroidal MR antagonist is tested for safety, efficacy and reduction of renal and cardiovascular morbidity and mortality in the ongoing phase 3 clinical trials: FINESSE-HF (EUCTR2015-002168-17), FIDELIO-DKD (NCT02540993) and FIGARO-DKD (NCT02545049), studies that will include over 14,000 participants (230-232).
Selective antagonists of 11β-HSD1 were also synthesized and tested in preclinical studies and in healthy human subjects. They show good safety profiles in clinical trials and seem to be particularly useful for treatment of Cushing syndrome, and metabolic syndrome characterized by obesity, dyslipidaemia, insulin resistance and hypertension. In addition to their metabolic effects, these inhibitors also show a modest blood pressure-lowering effect in T2DM patients and patients with obesity (233-236). Moreover, preclinical studies in mouse and rat models indicate that pharmacological inhibition of 11β-HSD1 confers protective effects against adverse cardiac remodelling (117, 237, 238). Nonetheless, efficacy of 11β-HSD1 inhibitors in preventing vascular and cardiac remodelling or reducing adverse cardiovascular outcomes has not been established yet in clinical trials (117, 239, 240).
Recently, inhibitors of aldosterone synthase (CYP11B2) have been developed and are currently screened for their therapeutic potential in cardiovascular diseases (239, 241). Some of these compounds have been tested in the clinical studies in healthy volunteers and in patients with primary aldosteronism (51, 242-245). Highly homologous structure of aldosterone (CYP11B2) and cortisol (CYP11B1) synthases render selective inhibition of aldosterone synthase difficult. Thus, the initial human trials showed that even though aldosterone synthase inhibitors, such as LCI699, are well tolerated and exert blood pressure-lowering effect, their low selectivity for aldosterone synthase leads to suppression of both aldosterone and glucocorticoids (243). However, recently published trials in healthy humans indicate that new highly selective inhibitors of aldosterone synthase – LY3045697 and RO683619 – potently blunt aldosterone release with insignificant effects on plasma concentrations of cortisol and ACTH, offering a potential advantage over less selective aldosterone synthase inhibitors previously evaluated in human trials. These new compounds exhibit significantly more potent and selective inhibition of CYP11B2 over CYP11B1. They are well tolerated by healthy subjects and their administration is not associated with significant side-effects, however multiple application of high doses can result in hyperkalaemia (244). Furthermore, new highly selective aldosterone synthase inhibitors with low affinity for CYP11B1 have been recently developed and successfully tested in monkeys (246).
SUMMARY
Mineralocorticoid receptors play a critical role in regulation and pathological remodelling of the cardiovascular system. MRs are expressed in organs involved in cardiovascular homeostasis: brain, heart, kidneys and vessels. Due to low selectivity, MRs bind both aldosterone and GCs and binding of GCs to MRs is largely determined by availability of tissue specific expression of 11β-HSD2, which converts GCs to inactive metabolites and allows for selective stimulation of MRs by aldosterone. 11β-HSD2 is expressed in the vascular wall, renal epithelium and some diencephalic and brain-stem nuclei involved in the regulation of water-electrolyte balance and blood pressure. In contrast, cardiac expression of 11β-HSD2 is low, thus, both aldosterone and GCs interact with cardiac MRs. As it is shown in Fig. 2, the excessive activation of MRs exerts several deleterious effects on the cardiovascular system, chiefly through sympatho-excitation, elevated salt appetite, renal retention of salt with consequent positive sodium balance, fibrosis and remodelling of the heart and arteries, as well as with propensity for atrial and ventricular arrhythmias. Furthermore, MR-mediated changes in the cardiovascular system are potentiated by RAS and activation of AT1Rs. The importance of MRs in the cardiovascular pathology is reflected in clinical guidelines that recommend use of MR blockers, spironolactone and eplerenone, in the treatment of heart failure, myocardial infarction and hypertension. Furthermore, new MR blockers, selective inhibitors of 11β-HSD1 and selective aldosterone synthase inhibitors have been developed and are currently tested in clinical trials.
Abbreviations: 11β-HSD1, 11β-hydroxysteroid dehydrogenase type 1; 11β-HSD2, 11β-hydroxysteroid dehydrogenase type 2; ACE, angiotensin converting enzyme; ACTH, adrenocorticotropic hormone; AF, atrial fibrillation; Ang II, angiotensin II; Ang III, angiotensin III; ARB, angiotensin receptor blocker; ARTS-HF study, The Mineralocorticoid Receptor antagonist Tolerability Study-Heart Failure; AT1R, angiotensin type 1 receptor; AT2R, angiotensin type 2 receptor; AVP, vasopressin; BMK1, Big MAP kinase1; BNP, brain natriuretic peptide; CBP, CREB-binding protein; CCR5, C-C chemokine receptor type 5; CKD, chronic kidney disease; CNS, central nervous system; cSrc, cytoplasmic tyrosine-protein kinase; CTGF, connective tissue growth factor; CYP11B2, aldosterone synthase (cytochrome P450 family 11 subfamily B member 2); DBD, DNA-binding domain; DOC, 11-deoxycorticosterone; ECM, extracellular matrix; EMPHASIS-HF, Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure; ENaC, epithelial sodium channel; EPHESUS, Eplerenone Post Acute Myocardial Infarction Heart Failure Efficacy and Survival Study; ER, endoplasmic reticulum; ERK, extracellular signal-regulated protein kinase; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; Flt1, FMS-like tyrosine kinase 1; GCs, glucocorticoids; GPCR, G-protein coupled receptor family; GRs, glucocorticoid receptors; HRE, hormone response element; HSP, heat shock protein; H6PDH, hexose-6-phosphate dehydrogenase; iNOS, inducible nitric oxide synthase; IP3, inositol triphosphate; Ito, transient outward potassium current; LBD, ligand binding domain; LVEF, left ventricular ejection fraction; MAPK, mitogen activated protein kinase; MCP, membrane cofactor protein-1; MDM2, mouse double minute 2 homolog; MEK, mitogen-activated protein kinase/ERK kinase; MI, myocardial infarction; MKP-1, MAPK phosphatase-1; MMP, matrix metalloproteinase; MRA, mineralocorticoid receptor antagonist; MRs, mineralocorticoid receptors; NCoR, nuclear receptor corepressor; NFκB - nuclear factor kappa B (nuclear factor κ-light-chain-enhancer of activated B cells); NHE-1, sodium hydrogen antiporter; NO, nitric oxide; NOX, NADPH oxidase; nSRE, negative steroid response element; NTD, N-terminal transactivation domain; NTS, nucleus of the solitary tract; NYHA, New York Heart Association; PAI-1, plasminogen activator inhibitor 1; PBMCs, peripheral blood mononuclear cells; PBP, peroxisome proliferator-activated receptor (PPAR)-binding protein; PGF, placental growth factor; PI3K, phosphoinositide 3-kinases; PKC, protein kinase C; PVN, hypothalamic paraventricular nucleus; p38MAPK, p38 mitogen-activated protein kinase; RAAS, renin-angiotensin-aldosterone system; Raf, family of serine/threonine protein kinases (acronym for rapidly accelerated fibrosarcoma); RALES, Randomized Aldactone Evaluation Study; Ras, family of GTP-binding proteins; RAS, renin-angiotensin system; ROS, reactive oxygen species; SFO, subfornical organ; SHR-SP, stroke-prone spontaneously hypertensive rat; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor; SON, supraoptic nucleus; SPIR-AF study, spironolactone-atrial fibrillation study; SRC, steroid receptor coactivator; TGF-β, transforming growth factor β; TIMP-1, tissue inhibitor of metalloproteinase 1; TRAP220, thyroid hormone receptor-associated protein (subunit 220); T2DM, type 2 diabetes mellitus; VEGFA, vascular endothelial growth factor A; VSMCs, vascular smooth muscle cells.
Acknowledgements: We thank Marcin Kumosa for his technical assistance in preparing figures.
The study was carried out with the use of CePT infrastructure financed by the European Union – the European Regional Development Fund within the Operational Programme “Innovative economy” for 2007-2013.
Conflict of interests: None declared.
REFERENCES
- Mangelis A, Juhlen R, Dieterich P, et al. A steady state system for in vitro evaluation of steroidogenic pathway dynamics: Application for CYP11B1, CYP11B2 and CYP17 inhibitors. J Steroid Biochem Mol Biol 2018; Dec 6: S0960-S0760. doi: 10.1016/j.jsbmb.2018.12.003.
- Grundy HM, Simpson SA, Tait JF. Isolation of a highly active mineralocorticoid from beef adrenal extract. Nature 1952; 169: 795-796.
- MacKenzie SM, Clark CJ, Fraser R, Gomez-Sanchez CE, Connell JM, Davies E. Expression of 11beta-hydroxylase and aldosterone synthase genes in the rat brain. J Mol Endocrinol 2000; 24: 321-328.
- Davies E, MacKenzie SM. Extra-adrenal production of corticosteroids. Clin Exp Pharmacol Physiol 2003; 30: 437-445.
- Tsybouleva N, Zhang L, Chen S, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 2004; 109: 1284-1291.
- Briones AM, Nguyen Dinh Cat A, Callera GE, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012; 59: 1069-1078.
- Yamamoto N, Yasue H, Mizuno Y, et al. Aldosterone is produced from ventricles in patients with essential hypertension. Hypertension 2002; 39: 958-962.
- Luther JM. Aldosterone in vascular and metabolic dysfunction. Curr Opin Nephrol Hypertens 2016; 25: 16-21.
- Vaidya A, Underwood PC, Hopkins PN, et al. Abnormal aldosterone physiology and cardiometabolic risk factors. Hypertension 2013; 61: 886-893.
- Gomez-Sanchez EP, Ahmad N, Romero DG, Gomez-Sanchez CE. Origin of aldosterone in the rat heart. Endocrinology 2004; 145: 4796-4802.
- Tsutamoto T, Wada A, Maeda K, et al. Transcardiac gradient of aldosterone before and after spironolactone in patients with congestive heart failure. J Cardiovasc Pharmacol 2003; 41 (Suppl. 1): S19-S22.
- MacKenzie SM, Connell JM, Davies E. Non-adrenal synthesis of aldosterone: a reality check. Mol Cell Endocrinol 2012; 350: 163-167.
- Bollag WB. Regulation of aldosterone synthesis and secretion. Compr Physiol 2014; 4: 1017-1055.
- Grazzini E, Boccara G, Joubert D, et al. Vasopressin regulates adrenal functions by acting through different vasopressin receptor subtypes. Adv Exp Med Biol 1998; 449: 325-334.
- Hinson JP, Kapas S, Teja R, Vinson GP. Effect of the endothelins on aldosterone secretion by rat zona glomerulosa cells in vitro. J Steroid Biochem Mol Biol 1991; 40: 437-439.
- Humphery TJ, Coghlan JP, Denton DA, et al. Effect of potassium, angiotensin II and ACTH on blood aldosterone and cortisol in sheep on different dietary potassium and sodium intakes. Clin Exp Pharmacol Physiol 1984; 11: 97-100.
- Kapas S, Purbrick A, Hinson JP. A ction of opioid peptides on the rat adrenal cortex: stimulation of steroid secretion through a specific mu opioid receptor. J Endocrinol 1995; 144: 503-510.
- Mazzocchi G, Malendowicz LK, Aragona F, Spinazzi R, Nussdorfer GG. Cholecystokinin (CCK) stimulates aldosterone secretion from human adrenocortical cells via CCK2 receptors coupled to the adenylate cyclase/protein kinase A signaling cascade. J Clin Endocrinol Metab 2004; 89: 1277-1284.
- Miura S, Nakayama A, Tomita S, Matsuo Y, Suematsu Y, Saku K. Comparison of aldosterone synthesis in adrenal cells, effect of various AT1 receptor blockers with or without atrial natriuretic peptide. Clin Exp Hypertens 2015; 37: 353-357.
- Rebuffat P, Malendowicz LK, Nussdorfer GG, Mazzocchi G. Stimulation of endogenous nitric oxide production is involved in the inhibitory effect of adrenomedullin on aldosterone secretion in the rat. Peptides 2001; 22: 923-926.
- Crabbe J. Site of action of aldosterone on the bladder of the toad. Nature 1963; 200: 787-788.
- Wright FS, Giebisch G. Renal potassium transport: contributions of individual nephron segments and populations. Am J Physiol 1978; 235: F515-F527.
- Pacha J, Frindt G, Antonian L, Silver RB, Palmer LG. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol 1993; 102: 25-42.
- Asher C, Wald H, Rossier BC, Garty H. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol 1996; 271: C605-C611.
- Frindt G, Sackin H, Palmer LG. Whole-cell currents in rat cortical collecting tubule: low-Na diet increases amiloride-sensitive conductance. Am J Physiol 1990; 258: F562-F567.
- Epstein AN. Mineralocorticoids and cerebral angiotensin may act together to produce sodium appetite. Peptides 1982; 3: 493-494.
- Shade RE, Blair-West JR, Carey KD, Madden LJ, Weisinger RS, Denton DA. Synergy between angiotensin and aldosterone in evoking sodium appetite in baboons. Am J Physiol Regul Integr Comp Physiol 2002; 283: R1070-R1078.
- Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB. Does aldosterone upregulate the brain renin-angiotensin system in rats with heart failure? Hypertension 2008; 51: 727-733.
- Gomez-Sanchez EP, Ahmad N, Romero DG, Gomez-Sanchez CE. Is aldosterone synthesized within the rat brain? Am J Physiol Endocrinol Metab 2005; 288: E342-E346.
- MacKenzie SM, Dewar D, Stewart W, Fraser R, Connell JM, Davies E. The transcription of steroidogenic genes in the human cerebellum and hippocampus: a comparative survey of normal and Alzheimer’s tissue. J Endocrinol 2008; 196: 123-130.
- Gomez-Sanchez EP. Brain mineralocorticoid receptors in cognition and cardiovascular homeostasis. Steroids 2014; 91: 20-31.
- Jackiewicz E, Szczepanska-Sadowska E, Maslinski W. Expression of mineralocorticoid receptors mRNA in the brain, heart and kidney of Sprague Dawley rats with renovascular hypertension. Brain Res Bull 2005; 65: 23-29.
- Teruyama R, Sakuraba M, Wilson LL, Wandrey NE, Armstrong WE. Epithelial Na(+) sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. Am J Physiol Endocrinol Metab 2012; 302: E273-E285.
- Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol 2005; 289: R1787-R1797.
- Han F, Ozawa H, Matsuda K, Nishi M, Kawata M. Colocalization of mineralocorticoid receptor and glucocorticoid receptor in the hippocampus and hypothalamus. Neurosci Res 2005; 51: 371-381.
- Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol 2009; 297: F559-F576.
- van Eekelen JA, Bohn MC, de Kloet ER. Postnatal ontogeny of mineralocorticoid and glucocorticoid receptor gene expression in regions of the rat tel- and diencephalon. Brain Res Dev Brain Res 1991; 61: 33-43.
- Young MJ, Rickard AJ. Mineralocorticoid receptors in the heart: lessons from cell-selective transgenic animals. J Endocrinol 2015; 224: R1-R13.
- Funder JW. Aldosterone and mineralocorticoid receptors-physiology and pathophysiology. Int J Mol Sci 2017; 18: 1032. doi: 10.3390/ijms18051032
- Milik E, Szczepanska-Sadowska E, Maslinski W, Cudnoch-Jedrzejewska A. Enhanced expression of mineralocorticoid receptors in the heart after the myocardial infarct in rats. J Physiol Pharmacol 2007; 58: 745-755.
- Kritis AA, Gouta CP, Liaretidou EI, Kallaras KI. Latest aspects of aldosterone actions on the heart muscle. J Physiol Pharmacol 2016; 67: 21-30.
- Mihailidou AS, Tzakos AG, Ashton AW. Non-genomic effects of aldosterone. Vitam Horm 2019; 109: 133-149.
- Ashton AW, Le TY, Gomez-Sanchez CE, et al. Role of nongenomic signaling pathways activated by aldosterone during cardiac reperfusion injury. Mol Endocrinol 2015; 29: 1144-1155.
- Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015; 65: 257-263.
- Jia G, Aroor AR, Sowers JR. The role of mineralocorticoid receptor signaling in the cross-talk between adipose tissue and the vascular wall. Cardiovasc Res 2017; 113: 1055-1063.
- Groeneweg FL, Karst H, de Kloet ER, Joels M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol 2012; 350: 299-309.
- Oakley RH, Cidlowski JA. Glucocorticoid signaling in the heart: a cardiomyocyte perspective. J Steroid Biochem Mol Biol 2015; 153: 27-34.
- Ruhs S, Nolze A, Hubschmann R, Grossmann C. 30 years of the mineralocorticoid receptor: nongenomic effects via the mineralocorticoid receptor. J Endocrinol 2017; 234: T107-T124.
- Kolkhof P, Barfacker L. 30 years of the mineralocorticoid receptor: mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol 2017; 234: T125-T140.
- Tesch GH, Young MJ. Mineralocorticoid receptor signaling as a therapeutic target for renal and cardiac fibrosis. Front Pharmacol 2017; 8: 313. doi: 10.3389/fphar.2017.00313
- Brem AS, Gong R. Therapeutic targeting of aldosterone: a novel approach to the treatment of glomerular disease. Clin Sci (Lond) 2015; 128: 527-535.
- Laursen SB, Finsen S, Marcussen N, Quaggin SE, Hansen PBL, Dimke H. Endothelial mineralocorticoid receptor ablation does not alter blood pressure, kidney function or renal vessel contractility. PLoS One 2018; 13: e0193032. doi: 10.1371/journal.pone.0193032
- Tarjus A, Belozertseva E, Louis H, et al. Role of smooth muscle cell mineralocorticoid receptor in vascular tone. Pflugers Arch 2015; 467: 1643-1650.
- Gueret A, Harouki N, Favre J, et al. Vascular smooth muscle mineralocorticoid receptor contributes to coronary and left ventricular dysfunction after myocardial infarction. Hypertension 2016; 67: 717-723.
- McCurley A, Pires PW, Bender SB, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 2012; 18: 1429-1433.
- Messaoudi S, Azibani F, Delcayre C, Jaisser F. Aldosterone, mineralocorticoid receptor, and heart failure. Mol Cell Endocrinol 2012; 350: 266-272.
- Fraccarollo D, Berger S, Galuppo P, et al. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 2011; 123: 400-408.
- Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol 2003; 284: H176-H184.
- Bienvenu LA, Morgan J, Rickard AJ, et al. Macrophage mineralocorticoid receptor signaling plays a key role in aldosterone-independent cardiac fibrosis. Endocrinology 2012; 153: 3416-3425.
- Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, Maeda N. Glucocorticoid- and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. Glia 1997; 20: 23-37.
- Le Menuet D, Lombes M. The neuronal mineralocorticoid receptor: from cell survival to neurogenesis. Steroids 2014; 91: 11-19.
- Chen J, Gomez-Sanchez CE, Penman A, May PJ, Gomez-Sanchez E. Expression of mineralocorticoid and glucocorticoid receptors in preautonomic neurons of the rat paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 2014; 306: R328-R340.
- Oyamada N, Sone M, Miyashita K, et al. The role of mineralocorticoid receptor expression in brain remodeling after cerebral ischemia. Endocrinology 2008; 149: 3764-3777.
- Gomez-Sanchez E, Gomez-Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol 2014; 4: 965-994.
- Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci USA 2001; 98: 5671-5676.
- Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med 1997; 48: 231-240.
- Essick EE, Sam F. Cardiac hypertrophy and fibrosis in the metabolic syndrome: a role for aldosterone and the mineralocorticoid receptor. Int J Hypertens 2011; 2011: 346985. doi: 10.4061/2011/346985
- Rentoukas EI, Lazaros GA, Zirogiannis PN. Aldosterone in heart and kidney diseases. Hellenic J Cardiol 2005; 46: 408-419.
- Wehling M. Rapid actions of aldosterone revisited: receptors in the limelight. J Steroid Biochem Mol Biol 2018; 176: 94-98.
- Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA 2005; 102: 19204-19207.
- Kanatsou S, Karst H, Kortesidou D, et al. Overexpression of mineralocorticoid receptors in the mouse forebrain partly alleviates the effects of chronic early life stress on spatial memory, neurogenesis and synaptic function in the dentate gyrus. Front Cell Neurosci 2017; 11: 132. doi: 10.3389/fncel.2017.00132
- Brailoiu GC, Benamar K, Arterburn JB, et al. Aldosterone increases cardiac vagal tone via G protein-coupled oestrogen receptor activation. J Physiol 2013; 591: 4223-4235.
- Qiao H, Hu B, Zhou H, et al. Aldosterone induces rapid sodium intake by a nongenomic mechanism in the nucleus tractus solitarius. Sci Rep 2016; 6: 38631. doi: 10.1038/srep38631
- Milik E, Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A, Dobruch J, Morton M, Koperski L. Upregulation of angiotensin AT1a receptors mRNA in the heart and renal medulla after myocardial infarction in rats. J Physiol Pharmacol 2006; 57: 375-388.
- Milik E, Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E. Effect of chronic mild stress on AT1 receptor messenger RNA expression in the brain and kidney of rats. Psychosom Med 2016; 78: 208-220.
- Di Zhang A, Nguyen Dinh Cat A, Soukaseum C, et al. Cross-talk between mineralocorticoid and angiotensin II signaling for cardiac remodeling. Hypertension 2008; 52: 1060-1067.
- Tsai CF, Yang SF, Chu HJ, Ueng KC. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int J Cardiol 2013; 169: 17-28.
- Zhang Z-H, Yu Y, Wei S-G, Felder RB. Aldosterone-induced brain MAPK signaling and sympathetic excitation are angiotensin II type-1 receptor dependent. Am J Physiol Heart Circ Physiol 2012; 302: H742-H751.
- Callera GE, Montezano AC, Yogi A, et al. c-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats. Hypertension 2005; 46: 1032-1038.
- Callera GE, Touyz RM, Tostes RC, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 2005; 45: 773-779.
- Grossmann C, Benesic A, Krug AW, et al. Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions. Mol Endocrinol 2005; 19: 1697-1710.
- Grossmann C, Freudinger R, Mildenberger S, Krug AW, Gekle M. Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. Am J Physiol Renal Physiol 2004; 286: F1226-F1231.
- Funder JW. Non-genomic actions of aldosterone: role in hypertension. Curr Opin Nephrol Hypertens 2001; 10: 227-230.
- Grossmann C, Gekle M. Non-classical actions of the mineralocorticoid receptor: misuse of EGF receptors? Mol Cell Endocrinol 2007; 277: 6-12.
- Min LJ, Mogi M, Li JM, Iwanami J, Iwai M, Horiuchi M. Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ Res 2005; 97: 434-442.
- Caldiz CI, Diaz RG, Nolly MB, et al. Mineralocorticoid receptor activation is crucial in the signalling pathway leading to the Anrep effect. J Physiol 2011; 589: 6051-6061.
- Harvey BJ, Thomas W. Aldosterone-induced protein kinase signalling and the control of electrolyte balance. Steroids 2018; 133: 67-74.
- Gao J, Zhang K, Chen J, et al. Roles of aldosterone in vascular calcification: An update. Eur J Pharmacol 2016; 786: 186-193.
- McEneaney V, Harvey BJ, Thomas W. Aldosterone regulates rapid trafficking of epithelial sodium channel subunits in renal cortical collecting duct cells via protein kinase D activation. Mol Endocrinol 2008; 22: 881-892.
- Kusche-Vihrog K, Callies C, Fels J, Oberleithner H. The epithelial sodium channel (ENaC): mediator of the aldosterone response in the vascular endothelium? Steroids 2010; 75: 544-549.
- Ferreira NS, Cau SB, Silva MA, et al. Diabetes impairs the vascular effects of aldosterone mediated by G protein-coupled estrogen receptor activation. Front Pharmacol 2015; 6: 34. doi: 10.3389/fphar.2015.00034
- Kolodziejczyk P, Gromotowicz-Poplawska A, Aleksiejczuk M, Chabielska E, Tutka P, Miltyk W. New sides of aldosterone action in cardiovascular system as potential targets for therapeutic intervention. Curr Drug Targets 2018; 19: 1968-1679.
- Hermidorff MM, de Assis LV, Isoldi MC. Genomic and rapid effects of aldosterone: what we know and do not know thus far. Heart Fail Rev 2017; 22: 65-89.
- Feldman RD, Limbird LE. GPER (GPR30): A nongenomic receptor (GPCR) for steroid hormones with implications for cardiovascular disease and cancer. Annu Rev Pharmacol Toxicol 2017; 57: 567-584.
- Beaumont K, Fanestil DD. Characterization of rat brain aldosterone receptors reveals high affinity for corticosterone. Endocrinology 1983; 113: 2043-2051.
- Krozowski ZS, Funder JW. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA 1983; 80: 6056-6060.
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 1985; 117: 2505-2511.
- Sheppard KE, Funder JW. Equivalent affinity of aldosterone and corticosterone for type I receptors in kidney and hippocampus: direct binding studies. J Steroid Biochem 1987; 28: 737-42.
- Haque M, Wilson R, Sharma K, Mills NJ, Teruyama R. Localisation of 11β-hydroxysteroid dehydrogenase type 2 in mineralocorticoid receptor expressing magnocellular neurosecretory neurones of the rat supraoptic and paraventricular nuclei. J Neuroendocrinol 2015; 27: 835-849.
- Cope CL, Black E. The production rate of cortisol in man. Br Med J 1958; 1: 1020-1024.
- Kotelevtsev Y, Holmes MC, Burchell A, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 1997; 94: 14924-14929.
- Stewart PM, Krozowski ZS. 11 beta-hydroxysteroid dehydrogenase. Vitam Horm 1999; 57: 249-324.
- Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001; 142: 1371-1376.
- Rajan V, Edwards CR, Seckl JR. 11 beta-hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J Neurosci 1996; 16: 65-70.
- Draper N, Stewart PM. 11beta-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol 2005; 186: 251-271.
- Agarwal AK, Mune T, Monder C, White PC. NAD(+)-dependent isoform of 11 beta-hydroxysteroid dehydrogenase. Cloning and characterization of cDNA from sheep kidney. J Biol Chem 1994; 269: 25959-25962.
- Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett 2004; 571: 129-133.
- Bujalska IJ, Draper N, Michailidou Z, et al. Hexose-6-phosphate dehydrogenase confers oxo-reductase activity upon 11 beta-hydroxysteroid dehydrogenase type 1. J Mol Endocrinol 2005; 34: 675-684.
- Gomez-Sanchez EP. The mammalian mineralocorticoid receptor: tying down a promiscuous receptor. Exp Physiol 2010; 95: 13-18.
- Gomez-Sanchez EP, Romero DG, de Rodriguez AF, Warden MP, Krozowski Z, Gomez-Sanchez CE. Hexose-6-phosphate dehydrogenase and 11beta-hydroxysteroid dehydrogenase-1 tissue distribution in the rat. Endocrinology 2008; 149: 525-533.
- Stewart PM, Murry BA, Mason JI. Human kidney 11 beta-hydroxysteroid dehydrogenase is a high affinity nicotinamide adenine dinucleotide-dependent enzyme and differs from the cloned type I isoform. J Clin Endocrinol Metab 1994; 79: 480-484.
- Edwards CR, Stewart PM, Burt D, et al. Localisation of 11 beta-hydroxysteroid dehydrogenase - tissue specific protector of the mineralocorticoid receptor. Lancet 1988; 2: 986-989.
- Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 1988; 242: 583-585.
- Smith RE, Little PJ, Maguire JA, Stein-Oakley AN, Krozowski ZS. Vascular localization of the 11 beta-hydroxysteroid dehydrogenase type II enzyme. Clin Exp Pharmacol Physiol 1996; 23: 549-551.
- Naray-Fejes-Toth A, Watlington CO, Fejes-Toth G. 11 beta-hydroxysteroid dehydrogenase activity in the renal target cells of aldosterone. Endocrinology 1991; 129: 17-21.
- Sheppard KE, Autelitano DJ. 11beta-hydroxysteroid dehydrogenase 1 transforms 11-dehydrocorticosterone into transcriptionally active glucocorticoid in neonatal rat heart. Endocrinology 2002; 143: 198-204.
- Gray GA, White CI, Castellan RF, McSweeney SJ, Chapman KE. Getting to the heart of intracellular glucocorticoid regeneration: 11beta-HSD1 in the myocardium. J Mol Endocrinol 2017; 58: R1-R13.
- Sato A, Funder JW. High glucose stimulates aldosterone-induced hypertrophy via type I mineralocorticoid receptors in neonatal rat cardiomyocytes. Endocrinology 1996; 137: 4145-4153.
- Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 1994; 93: 2578-2583.
- Young MJ, Funder JW. The renin-angiotensin-aldosterone system in experimental mineralocorticoid-salt-induced cardiac fibrosis. Am J Physiol 1996; 271: E883-E888.
- Funder JW. RALES, EPHESUS and redox. J Steroid Biochem Mol Biol 2005; 93: 121-125.
- Qin W, Rudolph AE, Bond BR, et al. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ Res 2003; 93: 69-76.
- Nagata K, Obata K, Xu J, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension 2006; 47: 656-664.
- Agarwal AK, Giacchetti G, Lavery G, et al. CA-Repeat polymorphism in intron 1 of HSD11B2: effects on gene expression and salt sensitivity. Hypertension 2000; 36: 187-194.
- Stewart PM, Krozowski ZS, Gupta A, et al. Hypertension in the syndrome of apparent mineralocorticoid excess due to mutation of the 11 beta-hydroxysteroid dehydrogenase type 2 gene. Lancet 1996; 347: 88-891.
- Friso S, Carvajal CA, Fardella CE, Olivieri O. Epigenetics and arterial hypertension: the challenge of emerging evidence. Transl Res 2015; 165: 154-165.
- Wang HW, Huang BS, Chen A, Ahmad M, White RA, Leenen FH. Role of brain aldosterone and mineralocorticoid receptors in aldosterone-salt hypertension in rats. Neuroscience 2016; 314: 90-105.
- Zhang ZH, Kang YM, Yu Y, et al. 11beta-hydroxysteroid dehydrogenase type 2 activity in hypothalamic paraventricular nucleus modulates sympathetic excitation. Hypertension 2006; 48: 127-133.
- Tsai CT, Chiang FT, Tseng CD, et al. Increased expression of mineralocorticoid receptor in human atrial fibrillation and a cellular model of atrial fibrillation. J Am Coll Cardiol 2010; 55: 758-770.
- Silvestre JS, Heymes C, Oubenaissa A, et al. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 1999; 99: 2694-2701.
- Yoshida M, Ma J, Tomita T, et al. Mineralocorticoid receptor is overexpressed in cardiomyocytes of patients with congestive heart failure. Congest Heart Fail 2005; 11: 12-16.
- Lavall D, Selzer C, Schuster P, et al. The mineralocorticoid receptor promotes fibrotic remodeling in atrial fibrillation. J Biol Chem 2014; 289: 6656-6668.
- Rocha R, Rudolph AE, Frierdich GE, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol 2002; 283: H1802-H1810.
- Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am J Pathol 2002; 161: 1773-1781.
- Smykiewicz P, Segiet A, Keag M, Zera T. Proinflammatory cytokines and ageing of the cardiovascular-renal system. Mech Ageing Dev 2018; 175: 35-45.
- Azibani F, Benard L, Schlossarek S, et al. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension 2012; 59: 1179-1187.
- Hori Y, Touei D, Saitoh R, et al. The aldosterone receptor antagonist eplerenone inhibits isoproterenol-induced collagen-I and 11beta-HSD1 expression in rat cardiac fibroblasts and the left ventricle. Biol Pharm Bull 2017; 40: 1716-1723.
- Zannad F, Dousset B, Alla F. Treatment of congestive heart failure: interfering the aldosterone-cardiac extracellular matrix relationship. Hypertension 2001; 38: 1227-1232.
- Favre J, Gao J, Zhang AD, et al. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am J Physiol Heart Circ Physiol 2011; 300: H2035-H2043.
- Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens 2004; 17: 597-603.
- Lijnen PJ, Petrov VV. Role of intracardiac renin-angiotensin-aldosterone system in extracellular matrix remodeling. Methods Find Exp Clin Pharmacol 2003; 25: 541-564.
- Robaczewska J, Kedziora-Kornatowska K, Kozakiewicz M, et al. Role of glutathione metabolism and glutathione-related antioxidant defense systems in hypertension. J Physiol Pharmacol 2016; 67: 331-337.
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev 1999; 79: 215-262.
- Ouvrard-Pascaud A, Sainte-Marie Y, Benitah JP, et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation 2005; 111: 3025-3033.
- Rossier MF, Lenglet S, Vetterli L, Python M, Maturana A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension 2008; 52: 721-728.
- Le Menuet D, Munier M, Meduri G, Viengchareun S, Lombes M. Mineralocorticoid receptor overexpression in embryonic stem cell-derived cardiomyocytes increases their beating frequency. Cardiovasc Res 2010; 87: 467-475.
- Lother A, Berger S, Gilsbach R, et al. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 2011; 57: 746-754.
- de Resende MM, Kauser K, Mill JG. Regulation of cardiac and renal mineralocorticoid receptor expression by captopril following myocardial infarction in rats. Life Sci 2006; 78: 3066-3073.
- Fraccarollo D, Galuppo P, Sieweke JT, Napp LC, Grobbecker P, Bauersachs J. Efficacy of mineralocorticoid receptor antagonism in the acute myocardial infarction phase: eplerenone versus spironolactone. ESC Heart Fail 2015; 2: 150-158.
- Rickard AJ, Morgan J, Bienvenu LA, et al. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012; 60: 1443-1450.
- Gerling IC, Ahokas RA, Kamalov G, et al. Gene expression profiles of peripheral blood mononuclear cells reveal transcriptional signatures as novel biomarkers of cardiac remodeling in rats with aldosteronism and hypertensive heart disease. JACC Heart Fail 2013; 1: 469-476.
- Kamalov G, Zhao W, Zhao T, et al. Atrophic cardiomyocyte signaling in hypertensive heart disease. J Cardiovasc Pharmacol 2013; 62: 497-506.
- Brand S, Amann K, Mandel P, Zimnol A, Schupp N. Oxidative DNA damage in kidneys and heart of hypertensive mice is prevented by blocking angiotensin II and aldosterone receptors. PLoS One 2014; 9: e115715. doi: 10.1371/journal.pone.0115715
- Konishi A, Tazawa C, Miki Y, et al. The possible roles of mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase type 2 in cardiac fibrosis in the spontaneously hypertensive rat. J Steroid Biochem Mol Biol 2003; 85: 439-442.
- Christ M, Douwes K, Eisen C, Bechtner G, Theisen K, Wehling M. Rapid effects of aldosterone on sodium transport in vascular smooth muscle cells. Hypertension 1995; 25: 117-123.
- Rautureau Y, Paradis P, Schiffrin EL. Cross-talk between aldosterone and angiotensin signaling in vascular smooth muscle cells. Steroids 2011; 76: 834-839.
- Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol 2016; 32: 659-668.
- Galmiche G, Pizard A, Gueret A, et al. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 2014; 63: 520-526.
- Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014; 63: 1033-1040.
- Wei Y, Whaley-Connell AT, Habibi J, et al. Mineralocorticoid receptor antagonism attenuates vascular apoptosis and injury via rescuing protein kinase B activation. Hypertension 2009; 53: 158-165.
- Ishizawa K, Izawa Y, Ito H, et al. Aldosterone stimulates vascular smooth muscle cell proliferation via big mitogen-activated protein kinase 1 activation. Hypertension 2005; 46: 1046-1052.
- Nakamura Y, Suzuki S, Suzuki T, et al. MDM2: a novel mineralocorticoid-responsive gene involved in aldosterone-induced human vascular structural remodeling. Am J Pathol 2006; 169: 362-371.
- Yan Y, Wang C, Lu Y, et al. Mineralocorticoid receptor antagonism protects the aorta from vascular smooth muscle cell proliferation and collagen deposition in a rat model of adrenal aldosterone-producing adenoma. J Physiol Biochem 2018; 74: 17-24.
- Hashimoto T, Ichiki T, Ikeda J, et al. Inhibition of MDM2 attenuates neointimal hyperplasia via suppression of vascular proliferation and inflammation. Cardiovasc Res 2011; 91: 711-719.
- Calvier L, Miana M, Reboul P, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol 2013; 33: 67-75.
- Jaffe IZ, Newfell BG, Aronovitz M, et al. Placental growth factor mediates aldosterone-dependent vascular injury in mice. J Clin Invest 2010; 120: 3891-3900.
- Harvey AP, Montezano AC, Hood KY, et al. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: the aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci 2017; 179: 110-119.
- Schafer A, Fraccarollo D, Hildemann S, et al. Inhibition of platelet activation in congestive heart failure by aldosterone receptor antagonism and ACE inhibition. Thromb Haemost 2003; 89: 1024-1030.
- Bodary PF, Sambaziotis C, Wickenheiser KJ, Rajagopalan S, Pitt B, Eitzman DT. Aldosterone promotes thrombosis formation after arterial injury in mice. Arterioscler Thromb Vasc Biol 2006; 26: 233. Doi: 10.1161/01.ATV.0000195782.07637.44
- Stankiewicz A, Gromotowicz A, Szemraj J, Wojewodzka-Zelezniakowicz M, Skrzypkowski P, Chabielska E. Acute aldosterone infusion enhances thrombosis development in normotensive rats. Thromb Haemost 2007; 98: 697-699.
- Gromotowicz A, Szemraj J, Stankiewicz A, et al. Study of the mechanisms of aldosterone prothrombotic effect in rats. J Renin Angiotensin Aldosterone Syst 2011; 12: 430-439.
- Gromotowicz-Poplawska A, Stankiewicz A, Mikita J, et al. Beneficial effect of combined spironolactone and quinapril treatment on thrombosis and hemostasis in 2K1C hypertensive rats. J Physiol Pharmacol 2018; 69: 245-253.
- Schafer A, Vogt C, Fraccarollo D, et al. Eplerenone improves vascular function and reduces platelet activation in diabetic rats. J Physiol Pharmacol 2010; 61: 45-52.
- Kramkowski K, Mogielnicki A, Buczko W. The physiological significance of the alternative pathways of angiotensin II production. J Physiol Pharmacol 2006; 57: 529-539.
- Billet S, Aguilar F, Baudry C, Clauser E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 2008; 74: 1379-1384.
- Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 2006; 20: 953-970.
- Sitia S, Tomasoni L, Atzeni F, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev 2010; 9: 830-834.
- Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Int Med 2003; 163: 1296-1304.
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007; 292: C82-C97.
- Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000; 105: 1605-1612.
- Ehsan A, McGraw AP, Aronovitz MJ, et al. Mineralocorticoid receptor antagonism inhibits vein graft remodeling in mice. J Thorac Cardiovasc Surg 2013; 145: 1642-1649.
- Rossi GP, Sacchetto A, Visentin P, et al. Changes in left ventricular anatomy and function in hypertension and primary aldosteronism. Hypertension 1996; 27: 1039-1045.
- Huang BS, Leenen FH. Mineralocorticoid actions in the brain and hypertension. Curr Hypertens Rep 2011; 13: 214-220.
- Dampney RA, Coleman MJ, Fontes MA, et al. Central mechanisms underlying short- and long-term regulation of the cardiovascular system. Clin Exp Pharmacol Physiol 2002; 29: 261-268.
- Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A, Ufnal M, Zera T. Brain and cardiovascular diseases: common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J Physiol Pharmacol 2010; 61: 509-521.
- Koneru B, Bathina CS, Cherry BH, Mifflin SW. Mineralocorticoid receptor in the NTS stimulates saline intake during fourth ventricular infusions of aldosterone. Am J Physiol Regul Integr Comp Physiol 2014; 306: R61-R66.
- de Kloet ER, Joels M. Brain mineralocorticoid receptor function in control of salt balance and stress-adaptation. Physiol Behav 2017; 178: 13-20.
- Zhang ZH, Yu Y, Kang YM, Wei SG, Felder RB. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. Am J Physiol Heart Circ Physiol 2008; 294: H1067-H1074.
- Lincevicius GS, Shimoura CG, Nishi EE, et al. Aldosterone contributes to sympathoexcitation in renovascular hypertension. Am J Hypertens 2015; 28: 1083-1090.
- Huang BS, Zheng H, Tan J, Patel KP, Leenen FH. Regulation of hypothalamic renin-angiotensin system and oxidative stress by aldosterone. Exp Physiol 2011; 96: 1028-1038.
- Leenen FH. The central role of the brain aldosterone- “ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta 2010; 1802: 1132-1139.
- Grassi G, Ram VS. Evidence for a critical role of the sympathetic nervous system in hypertension. J Am Soc Hypertens 2016; 10: 457-466.
- Grassi G, Seravalle G, Mancia G. Sympathetic activation in cardiovascular disease: evidence, clinical impact and therapeutic implications. Eur J Clin Invest 2015; 45: 1367-1375.
- Duprez DA, Bauwens FR, De Buyzere ML, et al. Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol 1993; 71: 17A-20A.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82: 1730-1736.
- Duprez DA, De Buyzere ML, De Backer T, et al. Influence of systemic arterial blood pressure and nonhemodynamic factors on the brachial artery pulsatility index in mild to moderate essential hypertension. Am J Cardiol 1993; 71: 350-353.
- Mizuno Y, Yoshimura M, Yasue H, et al. Aldosterone production is activated in failing ventricle in humans. Circulation 2001; 103: 72-77.
- Xue B, Beltz TG, Yu Y, et al. Central interactions of aldosterone and angiotensin II in aldosterone- and angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 2011; 300: H555-H564.
- Robert V, Heymes C, Silvestre JS, Sabri A, Swynghedauw B, Delcayre C. Angiotensin AT1 receptor subtype as a cardiac target of aldosterone: role in aldosterone-salt-induced fibrosis. Hypertension 1999; 33: 981-986.
- Belden Z, Deiuliis JA, Dobre M, Rajagopalan S. The role of the mineralocorticoid receptor in inflammation: focus on kidney and vasculature. Am J Nephrol 2017; 46: 298-314.
- Francis J, Weiss RM, Wei SG, et al. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol 2001; 281: H2241-H2251.
- Chen A, Huang BS, Wang HW, Ahmad M, Leenen FH. Knockdown of mineralocorticoid or angiotensin II type 1 receptor gene expression in the paraventricular nucleus prevents angiotensin II hypertension in rats. J Physiol 2014; 592: 3523-3536.
- Huang BS, White RA, Ahmad M, Leenen FH. Role of brain corticosterone and aldosterone in central angiotensin II-induced hypertension. Hypertension 2013; 62: 564-571.
- Huang BS, Chen A, Ahmad M, Wang HW, Leenen FH. Mineralocorticoid and AT1 receptors in the paraventricular nucleus contribute to sympathetic hyperactivity and cardiac dysfunction in rats post myocardial infarct. J Physiol 2014; 592: 3273-3286.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709-717.
- Bertocchio JP, Warnock DG, Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int 2011; 79: 1051-1060.
- Bosselmann H, Tonder N, Soletormos G, et al. Influence of renal impairment on aldosterone status, calcium metabolism, and vasopressin activity in outpatients with systolic heart failure. ESC Heart Fail 2017; 4: 554-562.
- Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther 2016; 161: 52-66.
- Kobayashi N, Hara K, Tojo A, et al. Eplerenone shows renoprotective effect by reducing LOX-1-mediated adhesion molecule, PKCepsilon-MAPK-p90RSK, and Rho-kinase pathway. Hypertension 2005; 45: 538-544.
- Bramlage P, Swift SL, Thoenes M, Minguet J, Ferrero C, Schmieder RE. Non-steroidal mineralocorticoid receptor antagonism for the treatment of cardiovascular and renal disease. Eur J Heart Fail 2016; 18: 28-37.
- Dojki FK, Bakris G. Nonsteroidal mineralocorticoid antagonists in diabetic kidney disease. Curr Opin Nephrol Hypertens 2017; 26: 368-374.
- Vizzardi E, Nodari S, Caretta G, et al. Effects of spironolactone on long-term mortality and morbidity in patients with heart failure and mild or no symptoms. Am J Med Sci 2014; 347: 271-276.
- Pitt B, Williams G, Remme W, et al. The EPHESUS trial: eplerenone in patients with heart failure due to systolic dysfunction complicating acute myocardial infarction. Eplerenone Post-AMI Heart Failure Efficacy and Survival Study. Cardiovasc Drugs Ther 2001; 15: 79-87.
- Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11-21.
- Tam TS, Wu MH, Masson SC, et al. Eplerenone for hypertension. Cochrane Database Syst Rev 2017; 2: CD008996.
- Ando K, Ohtsu H, Uchida S, et al. Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol 2014; 2: 944-953.
- Dudenbostel T, Calhoun DA. Use of aldosterone antagonists for treatment of uncontrolled resistant hypertension. Am J Hypertens 2017; 30: 103-109.
- Dabrowski R, Szwed H. Antiarrhythmic potential of aldosterone antagonists in atrial fibrillation. Cardiol J 2012; 19: 223-229.
- Dabrowski R, Borowiec A, Smolis-Bak E, et al. Effect of combined spironolactone-beta-blocker +/– enalapril treatment on occurrence of symptomatic atrial fibrillation episodes in patients with a history of paroxysmal atrial fibrillation (SPIR-AF study). Am J Cardiol 2010; 106: 1609-1614.
- Neefs J, van den Berg NW, Limpens J, et al. Aldosterone pathway blockade to prevent atrial fibrillation: a systematic review and meta-analysis. Int J Cardiol 2017; 231: 155-161.
- Sarwar CM, Papadimitriou L, Pitt B, et al. Hyperkalemia in heart failure. J Am Coll Cardiol 2016; 68: 1575-1589.
- Sarwar CMS, Bhagat AA, Anker SD, Butler J. Role of hyperkalemia in heart failure and the therapeutic use of potassium binders. Handb Exp Pharmacol 2017; 243: 537-560.
- Weir MR, Mayo MR, Garza D, et al. Effectiveness of patiromer in the treatment of hyperkalemia in chronic kidney disease patients with hypertension on diuretics. J Hypertens 2017; 35 (Suppl. 1): S57-S63.
- Danjuma MI, Mukherjee I, Makaronidis J, Osula S. Converging indications of aldosterone antagonists (spironolactone and eplerenone): a narrative review of safety profiles. Curr Hypertens Rep 2014; 16: 414. doi: 10.1007/s11906-013-0414-8
- Bakris GL, Agarwal R, Chan JC, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA 2015; 314: 884-894.
- Ruilope LM, Nowack C, Bakris GL. Masked and nocturnal hypertension in the ARTS-DN ABPM sub-study with finerenone. J Am Soc Hypertens 2016; 10 (Suppl. 1): e7. doi: 10.1016/j.jash.2016.06.021
- Pitt B, Anker SD, Bohm M, et al. Rationale and design of miner alocorticoid receptor antagonist tolerability study-heart failure (ARTS-HF): a randomized study of finerenone vs. eplerenone in patients who have worsening chronic heart failure with diabetes and/or chronic kidney disease. Eur J Heart Fail 2015; 17: 224-232.
- Ruilope LM, Agarwal R, Chan JC, et al. Rationale, design, and baseline characteristics of ARTS-DN: a randomized study to assess the safety and efficacy of finerenone in patients with type 2 diabetes mellitus and a clinical diagnosis of diabetic nephropathy. Am J Nephrol 2014; 40: 572-581.
- Filippatos G, Anker SD, Bohm M, et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J 2016; 37: 2105-2114.
- Pei H, Wang W, Zhao D, Wang L, Su GH, Zhao Z. The use of a novel non-steroidal mineralocorticoid receptor antagonist finerenone for the treatment of chronic heart failure: A systematic review and meta-analysis. Medicine (Baltimore) 2018; 97: e0254. doi: 10.1097/MD.0000000000010254
- Kolkhof P, Jaisser F, Kim SY, Filippatos G, Nowack C, Pitt B. Steroidal and novel non-steroidal mineralocorticoid receptor antagonists in heart failure and cardiorenal diseases: comparison at bench and bedside. Handb Exp Pharmacol 2017; 243: 271-305.
- Naegele M, Hernandez AF, Ruschitzka F. Finerenone in heart failure: walking a fine line. Eur Heart J 2016; 37: 2115-2117.
- Anderson A, Walker BR. 11beta-HSD1 inhibitors for the treatment of type 2 diabetes and cardiovascular disease. Drugs 2013; 73: 1385-1393.
- Shah S, Hermanowski-Vosatka A, Gibson K, et al. Efficacy and safety of the selective 11beta-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. J Am Soc Hypertens 2011; 5: 166-176.
- Freude S, Heise T, Woerle HJ, et al. Safety, pharmacokinetics and pharmacodynamics of BI 135585, a selective 11beta-hydroxysteroid dehydrogenase-1 (HSD1) inhibitor in humans: liver and adipose tissue 11beta-HSD1 inhibition after acute and multiple administrations over 2 weeks. Diabetes Obes Metab 2016; 18: 483-490.
- Feig PU, Shah S, Hermanowski-Vosatka A, et al. Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab 2011; 13: 498-504.
- Huang M, Liu J, Sheng Y, et al. 11beta-hydroxysteroid dehydrogenase type 1 inhibitor attenuates high-fat diet induced cardiomyopathy. J Mol Cell Cardiol 2018; 125: 106-116.
- Gordon O, He Z, Gilon D, et al. A transgenic platform for testing drugs intended for reversal of cardiac remodeling identifies a novel 11betaHSD1 inhibitor rescuing hypertrophy independently of re-vascularization. PLoS One 2014; 9: e92869. doi: 10.1371/journal.pone.0092869
- Monge M, Lorthioir A, Bobrie G, Azizi M. New drug therapies interfering with the renin-angiotensin-aldosterone system for resistant hypertension. J Renin Angiotensin Aldosterone Syst 2013; 14: 285-289.
- Wright DH, Stone JA, Crumley TM, et al. Pharmacokinetic-pharmacodynamic studies of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor MK-0916 in healthy subjects. Br J Clin Pharmacol 2013; 76: 917-931.
- Bernhardt R. The potential of targeting CYP11B. Expert Opin Ther Targets 2016; 20: 923-934.
- Azizi M, Amar L, Menard J. Aldosterone synthase inhibition in humans. Nephrol Dial Transplant 2013; 28: 36-43.
- Wang HZ, Tian JB, Yang KH. Efficacy and safety of LCI699 for hypertension: a meta-analysis of randomized controlled trials and systematic review. Eur Rev Med Pharmacol Sci 2015; 19: 296-304.
- Sloan-Lancaster J, Raddad E, Flynt A, Jin Y, Voelker J, Miller JW. LY3045697: Results from two randomized clinical trials of a novel inhibitor of aldosterone synthase. J Renin Angiotensin Aldosterone Syst 2017; 18: 1470320317717883. doi: 10.1177/1470320317717883
- Bogman K, Schwab D, Delporte ML, et al. Preclinical and early clinical profile of a highly selective and potent oral inhibitor of aldosterone synthase (CYP11B2). Hypertension 2017; 69: 189-196.
- Sakakibara R, Sasaki W, Onda Y, et al. Discovery of novel pyrazole-based selective aldosterone synthase (CYP11B2) inhibitors: a new template to coordinate the heme-iron motif of CYP11B2. J Med Chem 2018; 61: 5594-5608.
A c c e p t e d : February 28, 2019