ACRYLAMIDE TOXICITY AND CHOLINERGIC NERVOUS SYSTEM
INTRODUCTION
Despite many years of research, acrylamide (ACR) has an influence on the central-peripheral distal axonopathy and remains poorly understood. Based on accumulating evidence, it is possible that the disorder of elemental homeostasis represents an important component of the mechanism of ACR’s neurotoxicity. The mechanism of ACR’s neurotoxicity may be related to an impaired cholinergic transmission in the central and peripheral nervous system and a redox imbalance. These may not only affect ongoing brain functions but also participate in the etiology of neurodegeneration. This review summarizes our current knowledge (Fig. 1) of the relationship between acrylamide toxicity and central nervous system.
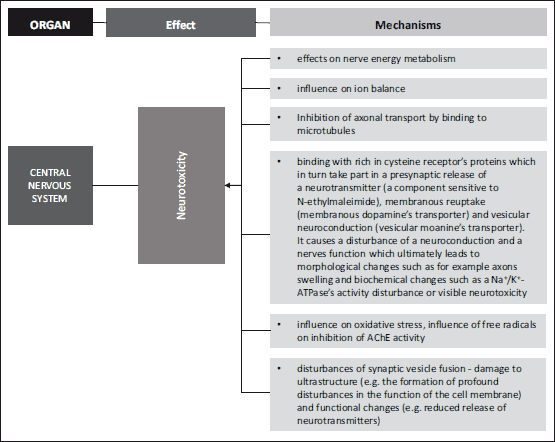 |
Fig. 1. Mechanisms leading to neurotoxicity (28, 29, 30, 33-44, 90-94, 116). |
ACRYLAMIDE AS A TOXIC SUBSTANCE IN FOOD
Acrylamide (H2C=CH-CONH2) is an organic compound with a low molecular weight (71.08 g) composed of carbon atoms (50.69 %), hydrogen (7.09%), nitrogen (19.71%), oxygen (22.51%).
In its original state, it is found to be a white, odourless and crystalline substance, with a melting point of 84.5ºC and 1.122g/cm3 of density in 30ºC. Due to its relatively low volatility, its boiling point equals 192.6ºC under 1 atm (101.3kPa) pressure. This chemical’s nature is polar due to the presence of functional groups. Furthermore, it is freely soluble in water in addition to the polar solvents, namely methanol or ethanol, while it is completely insoluble in petroleum benzine and heptane (1). Acrylamide, as an organic compound, is quite reactive, having conjugated dual binding and a part of amidein its structure. Its high chemical activity is mainly due to the presence of multiple binding with electrophilic properties. The double binding acts as the electrophilic center which easily binds with amino groups (-NH2) as well as with sulfhydryl’s amino acids (-SH), peptides and proteins. Furthermore, ACR is able to create some degree of hydrogen binding through the presence of the urea nitrogen group (2). Acrylamide reacts in various ways with some organic compounds with the amido and vinyl groups participate. These are, among others, the nucleophilic addition or Dielsa-Adler’s reactions. Like other vinyl group’s, ACR could be responsive to Michel’s addition (3). The above-mentioned compound demonstrates low acidity as well as low alkalinity properties. Due to the amide moieties, ACR reacts in the processes such as hydrolysis, dehydration, alcoholysis and the reaction of aldehyde’s condensation (4). Acrylamide undergoes the ultraviolet radiation, and polymerizes. It is found to be stable kept at room temperature until removed in a cool, dark place.
This organic compound is formed not only during the heat treatment of food products containing starch and carbohydrates, but also during high temperature meat frying (5-7).
The research into mechanisms of ACR’s formation entered a new era when it was discovered that it is formed when asparagina’s amino acid’s reacts with reducing saccharides (7). It demonstrated that ACR can be found in food consisting of the nitrogen or oil compounds which is heat-treated at 120ºC. The most probable process, which leads to the ACR’s formation, is associated with the production of acrolein, which is produced as a result of glycerol’s thermal decomposition followed by acrolein’s oxidation with acrylic acid. The acrylic and acrolein can be produced from fat released triglycerides, which are composed from food frying processes (6, 7). Furthermore, The International Agency for Research on Cancer (IARC) has classified ACR into 2A groups as a carcinogenic and mutagenic substance (8, 9). The carcinogenic potential is demonstrated only in the monomer’s form and never in the polymer (10). In 2002, The Swedish Food Administration (SFA) took a closer look at the extremely high ACR’s content in many food products (6). It was pointed out that ACR is also found in coffee. The highest mean of ACR’s concentrations were found in coffee substitutes whilst the lowest were for roasted coffee (11-14). A monitoring study throughout Europe during 2007 – 2009 showed that instant coffee may contain up to 4300 µg/kg of ACR (15). The ACR’s content in some food and drinks is summarized in Table 1. Prior research has shown that ACR may have the full capacity to create hemoglobine’s adducts – the compounds created out of an ACR and hemoglobin’s relation. It is the reason why they may be used as compounds exposition’s biomarkers. The SFA and the Stockholm University attested that the highest concentration of ACR can be found in some deep-fried or just fried products.
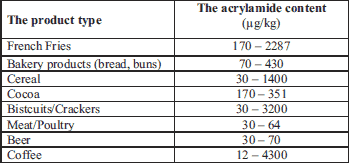
As for now, a maximal limit for the ACR’s contents in food products has not yet been specified. In accordance with the United Union’s provisions, potable water’s maximum allowable concentration of ACR is 0.1 µg/dm3. However, the Environmental Protection Agency (EPA) guidelines determine the amount to 0.5 µg/dm3 (16). The maximal ACR’s residuum in the case of cosmetics, shall not exceed 100 µg/kg (17). Whereas, the allowable ACR’s concentration in the production plant’s air shall not exceed 30 µg/m3 (1).
THE ACRYLAMIDE’S METABOLISM
Many researchers have been investigating the process of ACR formation during thermal processing of carbohydrated food at temperatures exceeding 120ºC. They described a hypothetical mechanism in which this compound could arise, mainly due to some reactions occurring among amino acids and reducing sugars in heated food (18). The hypothesis is based on the assumption that Millard’s reactions, which result in the food’s taste, smell and color, which are linked to a formation of ACR out of asparagine and reducing sugars (19, 20). Based on studies conducted by Stadler et al. (21), it was concluded, N-glycosides created as a result of the asparagine’s reaction with reducing sugars which lead to obtaining a considerate amount of ACR, whilst reactions of glutamine and methionine produced a much lower concentration of this compound. Implemented experiments confirm the asparagine’s key role in the process of ACR’s creation. The development of acrylamide from asparagine is connected to the Millard’s reaction, including a series of reactions leading towards various reactive intermediate products (3).
Based on the conducted rodent experiments, it was demonstrated that ACR, which gets to the organism, not only by an oral ingestion or airways, but also through the skin, is being is metabolized and excreted with the urine. The ACR fate in the organism is much more complex (22). The biotransformation and elimination of this compound occurs in the liver. With the participation of cytochrome P450 2E1, ACR is converted to an epoxide form – glycidamide (16). P450 cytochrome (CYP) belongs to the hemoprotein’s family. They play an important role in the bioactivation and detoxification of many hazardous substances. It is assumed that CYP1A and CYP2E mainly metabolized the carcinogenic substances, whilst CYP3A, CYP2D and CYP2C are mainly responsible for metabolizing drugs (23).
The acrylamide and glycidamide conjugate reactions with glutathione (GSH). As a result of the direct conjugation reaction of ACR with glutathione N-acetyl-S-(3-amino-3-oxypropyl), cysteine is produced. Glycidamide could conjugate with GSH involving the GST enzymes to N-acetyl-S- (2-carbamoyl-2-hydroxyethyl) cysteine and N-acetyl-S- (3-amino-2-hydroxy-3-oxopropyl) cysteines as well as enzymatic hydrolysis catalysed by the epoxide hydrolase to dihydroxypropanamide (3).
Sorgel et al. (24) demonstrated the ACR half-life period in an organism of 18 – 52 year-old men lasts 2 – 7 hours. It may be the characteristic of the relatively slow elimination (excretion) of this substance. It was demonstrated that the largest part of the ACR is metabolized (90%), while only a small amount of it is excreted in urine (25).
In relation to DNA, ACR demonstrates low reactivity. The glycidamide, in turn, as a more reactive compound, has the ability to create adduct out of DNA, which are created as a result of Michael’s nucleophilic addictions. This is why the glycidamide is regarded as being genotoxic. It can cause gene mutations and chromosome defects (13, 26). Until now, the obtained results of epidemiological studies have not provided inconclusive proof, which would confirm the interdependency between the ACR’s food intake and increased risk of cancer related morbidity. However, some research on experimental animals have demonstrated this chemical compound is carcinogenic for many organs such as the thyroid, testicles, lungs or skin (27). Quantitatively translating or comparing the research results of ACR’s carcinogenic effects on animals with humans is a rather difficult task for there are some very rodent specific factors which influence cancerous growths in these animals. On the other hand, there is some credible scientific evidence confirming the ACR’s neurotoxic influence on human body.
THE ACRYLAMIDE’S NEUROTOXIC INFLUENCE
Both glycidamide and acrylamide show neurotoxic activities (28). ACR administrated orally, intraperitoneally or by inhalation is found to be toxic. The compound’s toxicity screenings were run both on animal and human models by assessing the generated amount of adducts with hemoglobin. The amount of DL50 and CL50 acrylamide set for lab animals is shown in the Table 2.
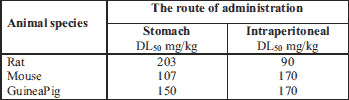
Symptoms of neurotoxicity were observed during a high dose (≥ 100 mg/kg b.w.) ACR’s administration. Fullerton and Barnes (29) carried out an experiment where the Porton’s rat group received a single 100 or 203 mg/kg b.w. (DL50 for female) dose of ACR, and was administrated via the stomach. In the animals which received the 203 mg/kg of ACR’s dose, slight tremors were observed, which lasted about 48 hours. It was subsequently observed that the rats either quickly recovered (2 or 3 days) or died. The slight tremor in rats was also caused by administrating a single 100 mg/kg ACR’s dose. By administration of the subsequent dose during a 24 hour period resulted in general weakness and death within three days. The persistent exposure to ACR’s influence can cause damage to the central and peripheral nervous system in animals as well as in humans. Human’s prodromal signs of such a conditioninclude numbness, tingling and ataxia (17, 30). This is linked to the damaging of the nervous system which might result in the inhibition of both the axonal transport and neurotransmission (27).
It would seem essential to understand the neurotoxicity’s mechanism, for such a system has been observed not only in animals but also in the ACR’s exposed population. There was an accident of ACR and N-methylacrylamide’s leakage in 1997 in Sweden, which happened during the construction of a tunnel. Acrylamide neurotoxic influence on humans was confirmed at that time (27). Neurological observations were made of distal axons edema and degeneration of axons in the central and peripheral nervous system. This was a distinctive neuropathological characteristic change caused by ACR. This has resulted in the qualification of an ACR caused neuropathy as a centrally-peripheral axonopathy. Nowadays, it is considered that nerve endings, and not axons, are the primary location of the ACR’s activity. The research results, which were obtained out of animal experiments, could as certain the early and progressive degeneration of nerve endings in all of the central nervous system (CNS) areas as well as the cerebellum’s damage of Purkinje cells. Furthermore, it has been assessed that axons degeneration was a secondary process (30, 31).
The brain and the great sciatic nerve’s creatine phosphokinase (CPK) are characterized by a particular ACR’s sensitivity which blocks its activity (32). The result of this enzyme’s inhibition is adenosine-5'-triphosphate (ATP) deficiency in a cell which as a consequence can result in apoptosis (33). It is worth noting that some experimental studies proved the CPK activity in a human’s brain is entirely inhibited by ACR. It is a proof that human’s brain is extremely sensitive to ACR (32). The prolonged contact with ACR may result in an irreversible CNS’s damage through neuronal impulse inhibition (30).
Research has also been conducted into worker’s ACR’s exposure in the work place. Workers, with an occupational exposure on ACR at least of 0.3 mg/m3 air dose, were observed to suffer from numbness of the arms and feet, as opposed to workers with less than 0.3 mg/m3 air dose exposition on ACR (27). Acrylamide’s necessary maximum concentration levels (NDS) is 0.05 mg/m3 of air. The binding occupational exposure limit values (BOELVs) in EU Member States play an important role. The European Advisory Committee on Safety and Health in the workplace (ACSH) has adapted a legislative of BOELV’s value of ACR’s concentration as of 0.07 ÷ 0.1 mg/m3 air. Taking into consideration the above findings, the ACR air value equals 0.07 mg/m3. It is worth highlighting that conducted studies of worker’s ACR’s exposure in the work place demonstrated a clear relationship between the level of ACR’s adducts with hemoglobin (N-(2-carbamoylethyl)-valine, AA-Hb) and the peripheral nervous system’s symptoms occurrence (34). The lowest concentration causing a biological effect – 0.51 nmol AA-Hb/g value was set for some earliest symptoms of tingling or numbness of extremities. The above-mentioned value corresponds to ACR 0.1 mg/m3 air concentration. This is Poland’s prevailing ACR value at NDS (27).
ACRYLAMIDE AND THE OXIDATIVE STRESS
The oxidative stress is commonly defined as a state in which reactive oxygen species (ROS) creation far exceeds the antioxidants protection ability. As demonstrated based on lab animal tests, that even small amounts of ACR cause different organs oxidative stress (28, 35). The organism’s oxidative protection occurs in three stages. Antioxidative enzymes, which function as a protection from free radicals, take part in a first phase. Scavengers which break free a radicals chain reaction, play the main role in a second phase. Subsequently, there is a recovery of ROS reaction with cells components in the third phase. In addition, the regeneration of healthy cells structure takes place (36).
Reactive oxygen species are created in many physiological processes. This caused the organism to produce, so-called antioxidant defense system (ADS) which protects against its harmful effects. The antioxidant defense system plays a very important role, which is to prevent the initiation of the oxidation reaction, as well as to repair the prior damage. The antioxidant defense system is comprised of free radical scavengers, antioxidant enzymes and preventative antioxidants. The production of free radicals is widely limited due to the valuable system’s performance. Meanwhile, superoxide anion, hydroxyl radicals or hydroperoxide radicals are transformed to O2 or H2O. The non-enzymatic system, which protects against an oxidation damage, is comprised of ascorbic acid, tocopherol, β-carotene, flavonoids and creatinine. Endogenous compounds are comprised of reduced GSH. This antioxidant serves many important roles, among others it neutralizes ROS, which in turn protects some reactive protein groups from an irreversible inactivation (37).
The reduced GSH to oxidized glutathione’s ratio is 10:1 in the correct cytosol as well as in mitochondrion of healthy cells. The intracellular GSH’s concentration depends on the cell’s type and it varies between 5 – 10 mmol/l. The high GSH’s concentration is found in the cytoplasm, the nucleus and in mitochondrion. Whilst a lower 2 mmol/l concentration was found in endoplasmic reticulum (38). The reduced GSH is regarded as the most important thiol buffer where the reduced to oxidized form ratio (glutathione/glutathione disulfide) is considered the oxidative-reductive state’s measure. There is a reaction between GSH and hydrogen peroxide at the time of the oxidative stress. The above-mentioned reaction is catalyzed by the glutathione’s peroxidase. The rise of glutathione disulfide’s concentration is the result of such a reaction. The GSH’s oxidized form is subsequently transformed into a GSH’s reduced form. The reaction is catalyzed by the GSH’s reductase. The GSH most important role is to keep thiol groups in a reduced form which is a condition for their functional activity. GSH has the capacity to reduce peroxides as well as to keep protein’s SH - group amount at a correct level. This compound has been recognized as one of the most important elements of the cell’s antioxidant system (39). Glutathione plays the role of a sweeper for both reactive oxygen species and electrophilic compounds. Its participation in a signal transductiongene expression or in an apoptosis is also regarded as a highly important function (40).
Glutathione constitutes the third line of defense. It takes part in a damaged cell’s components repair (40). GSH undergoes a reaction of conjugation with some toxic compounds such as, for instance, acrylamide. It is the reason for lowering its cells concentration. The reduction of brain and muscles GSH’s concentration may result from its consumption caused by reactions of free radicals created in an excessive amount after ACR’s administration (Fig. 2). Therefore, there may be danger of a serious oxidative-reductive imbalance (41, 42).
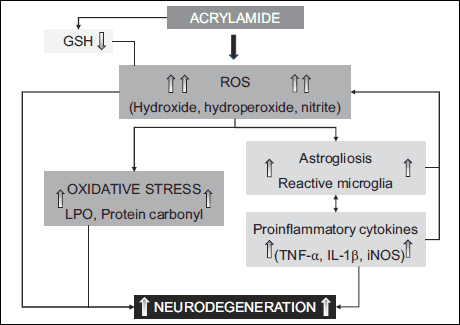 |
Fig. 2. The effect of acrylamide on oxidative stress (41). |
The organism’s oxidative capacity also depends on the content and activity of antioxidant proteins. Preventive antioxidants guard not only from the forming of new ROS but also from the beginning of the lipids peroxidation’s process. The albumins function, among others, as proteins which connect with the freed ions of transitions metals, such as copper or iron which in turn contain unpaired electrons functioning as protection against free radical reactions (36, 41).In vitro studies showed albumin protects erythrocytes against copper ions influenced peroxidation. It is mediated via ion binding. This prevents from the development of hydrogen peroxide created out of hydroxyl radicals (36).
One of the effects of oxidative stress is the lipids peroxidation process (36). Peroxidation is the oxidation process of unsaturated fatty acids which are part of phospholipids thus, as a consequence, the above-mentioned peroxides are created. The main product of lipid’s peroxidation is a malondialdehyde (MDA). The development of ROS in an organism results in MDA’s concentration raise. Consequently, a cell membrane’s permeability changes and mitochondrion’s oxidative phosphorylation uncouples, which may lead to an apoptosis as a result (42-44). Oxidation reactions related to acrylamide’s toxic influence contribute to lipids peroxidation and to a decline of a GSH’s concentration. A phenomena such as this may stimulate the increase of the enzyme’s activity for cascade conversions of arachidonic acid: cyclo-oxygenase and lipo-oxygenase. And moreover, this also causes calcium ions influx to the cell, which leads not only to the cell’s damage but also to the damage of its receptors. The consequence of oxidative stress is a cytoplasmic membrane’s depolarization, which in turn causes its raise in permeability. It results in a potential difference’s reduction between cell’s inside and outside environment (44, 45).
In an ADS’s structure the enzymatic system is created by specialized enzymes which remove free radicals as well as prevent their formation. The following enzymes can be found in the above-mentioned group, among others: catalase, glutathione peroxidase as well as superoxide dismutase. As a lot of research demonstrates (46-48), acrylamide reduces the above-mentioned enzyme’s activity which probably points to an oxidative-reductive balance disorder caused by its influence.
CHARACTERISTICS OF THE CHOLINERGIC SYSTEM
The cholinergic system is a subject of wide interest due to its role in an etiopathogenesis as well as in Alzheimer’s pharmacotherapy. The amount of scientific research into the cholinergic system has risen recently, mainly due to the cholinergic agonists possible therapeutic benefits in many neurodegenerative diseases. It has been demonstrated that in the above-mentioned disease there is a decrease in the number of the muscarinic and nicotinic receptors in the CNS (49, 50). The way how many neurotoxins work is related to their influence on the neurotransmission. Even the smallest changes in neurotransmitters system will have a substantial influence on brain’s bioelectric activity as well as in behavior. This is related to the interactions between certain neurotransmitters and neurons.
Acetylcholine (ACh) is the main cholinergic system’s transmitter. This is an acetic acid and choline’s ester. It is formed with a participation of choline’s acetyltransferase (51). Choline is transported with blood to the central nervous system, where it passes to neurons through an active transportation mode. It is synthesized in the neurons on a minimal scale. As research demonstrates, the stronger the cholinergic activity is, the more choline is taken from the blood via the nervous system (52). Acetylcholine is stored in synaptic vesicles and it is released to a synaptic space as a result of cell membrane’s depolarization.
Acetylcholine’s catabolism is caused by acetylcholinesterase (AChE) and butyrylcholinesterase which are responsible for hydrolysis reactions. Acetylcholinesterase is located in the central nervous system. Its high activity is also shown in muscles. Butyrylcholinesterase, on the other hand, can be found in tissues and peripheral organs including the liver. The butyrylcholinesterase’s presence has been established in glia (53). Choline is created as a result of the above-mentioned enzyme. It is reflexively absorbed to a neuron due to a high activity of choline’s transporter (54). There are muscarinic and nicotinic receptors within a cholinic system. Five types of muscarinic receptors have been separated. They have a connection with the G protein and they have seven heliacal transmembrane domains (55). Depending on the mechanism of a transduction signaling, two receptors families have been distinguished. The first group consists of the following receptors: M1, M3, M5. They activate a Gq protein and in this way they start a cascade of intracellular reactions of phosphatidylinositol.
The second group consists of M2 and M4 receptors which are connected with Gi and Go proteins. There is a blocking of cyclic adenosine monophosphate after their activation. The above-mentioned receptors often act as auto-receptors and its stimulation blocks the release of neurotransmitters form the neurons (52).
The central nervous system consists of nicotinic receptors in hippocampus, cortex, thalamus, hypothalamus, striatum, cerebellum, black substance, raphe nucleus and tegmentum. Nicotinic peripheral receptors are mainly located in skeletal muscles, adrenal glands and autonomic ganglia (52). The reduction in the number of nicotinic receptors in the cortex and hippocampus can be observed when faced with neurodegenerative diseases (54). Additionally, galantamine as an AChE’s inhibitor, modulates the nicotinic receptor’s capabilities thereby correcting cognitive functions (54-57). Cholinergic neurons may serve as interneurons andmay create longer routes enabling projections to a number of structures in CNS. The above-mentioned routes begin in the two centers. They reach upwardly to the hypothalamus, basic forebrain, thalamus, hippocampus, amygdala, the black substance and tegmentum ganglia from brain stem’s center. There are also cholinergic fibers, which descend to the cerebellum, medulla oblongata and cranial nerve’s nucleus. It is worth mentioning that cholinergic neurons also reach to the nucleus of locus coeruleus. This points to some direct interactions in the noradrenergic and cholinergic systems. The other significant structure, out of which cholinergic neurons derive, is the basic forebrain. The Meynert’s nucleus and septum’s central nucleus is located there. Cholinergic fibers reach the amygdala and hippocampus from there. It is a consistent fact, that cholinergic fibers reach the cortex, amygdala, frontal gyrus cinguli and olfactory bulb (57). There is a cholinergic innervation’s impoverishment in case of cholinergic fibers degenerative process in a prosencephalon’s nucleus. This is one of the Alzheimer’s etiopathogenesis explaining theories. The deepening deficit of cognitive functions correlates to the degree of cholinergic nerves loss (58, 59).
Choline acetyltransferase is mainly located in the cystole of cholinergic neuron terminations. However, a certain enzyme pool is connected with some membranes of the synaptic vesicles and cytomembranes. Such a position may be a cause of choline synthesis after the nerve endings penetration, thus a newly formed ACh may be immediately transported into the synaptic vesicles (60). The ACh’s synthesis depends on the substrate supply especially on choline (61). When choline’s blood levels increases, the ACh’s level off and release extending equally.
There are two similar brain systems which transport choline via cytomembranes. Almost half of the choline which was used to synthesis of ACh, comes out of a disintegrated ACh which was released to a synaptic gap (62, 63).
The main source of an acetyl-coenzyme A (acetyl-CoA), which is indispensable to ACh, synthesis is acetyl-CoA being produced in mitochondria out of pyruvate during a reaction catalyzed by pyruvate dehydrogenases (60). Due to various pathogenic factors, acetyl-CoA’s delivery disturbance may occur, where acetyl-CoA is used not only to enable energy’s production and structural lipid’s synthesis, but also to aid ACh’s production. This is the common cause of a selective cholinergic neurons’ degeneration (64). ACh has two active centers in its structure, enabling a connection with receptors, in other words, quaternary, alkaline nitrogen atom and an ester linkage. It has the ability to take different conformational forms which contributes to nicotinic and muscarinic receptor stimulation. ACh exists in a synclinal form which is energetically constant as and in an elongated form which is less permanent (65). Most of the ACh’s effects in the brain occur as a result of numerous muscarinic receptors stimulation. Neuronal nicotinic acetylcholine receptors (nAChR) are characterized by some specific pharmacological characteristics. The chronic agonist’s (nicotine) activity causes the doubling of its density (61). This receptor is a subject to some adaptive changes which lead to the sustaining of a transmission, thus avoiding the results of an AChR’s desensitization, which occurs as a result of a chronic exposition to agonist’s activity. Receptors are inactive even though they are in greater numbers or in other words, cations do not pass through the receptor’s canal (66). There are also presynaptic nAChR receptors varying widely in subunits composition. They increase the release of neurotransmitters form nerve endings. This release is dependent on Ca2+ ions (67).
THE BIOSYNTHESIS, THE RELEASE AND THE DEGENERATION OF ACETYLCHOLINESTERASE
Acetylcholine’s activity comes to an end in the time of its enzymatic disintegration in an intercellular space. It happens immediately after its cholinergic endings secretion via AChE (E.C.3.l.l.7.) also known as a specific or true choline’s esterase (68). It is one of the most active enzymes as well as a stable and a multiform compound. There are a few AChE’s forms known, these are globular as well as asymmetric forms. There are only globular forms in a tetrametric form occurring in the brain (69). Neurons, which synthesize AChE capable of its secretion, on which speed depends on many factors (mainly on neurotransmitter’s concentration change in a synaptic gap as well as on pharmacological products). The sudden AChE’s rise within a neuron’s vicinity may occur during that time. The secreted AChE may hydrolyze diffusing in intercellular space ACh is much more effective in comparison with the one connected with the membranes (68). Different AChE’s forms have similar catalictic properties even though they differ in properties and structure. Acetylcholinesterase is particularly responsive to organophosphorus compounds (irreversible inhibitors) and other inhibitors (reversible inhibitors), within permanent bonds (70). AChE’s activity is suppressed by the substrate’s excess. There is an ACh pool’s enhancement after the blocking of AChE’s activity.
Acetylcholinesterase also occurs in the brain’s spheres without cholinergic innervation. It may be indicative of its broad spectrum of activity. There was research conducted into the confirmation ofits function in the adhesion and morphogenesis processes (62).
Esterase acetylcholine hydrolysis acetylcholine into an acetic acid. This esterase breaks down only ACh. It can be found in the peripheral and the CNS, in erythrocytes, marrow, the spleen and lymph nodes (71). AChE by means of an immediate ACh’s break down doesn’t lead to its excessive collecting in synaptic gaps because this might cause disturbances in the correct functioning of an organism (67). ACh can be found in an excessive concentration in all the places where ACh plays the role of a nervous transmitter. Choline is reabsorbed into a presynaptic part on the principle of an active transport which requires the presence of sodium ions. Its function is to resynthesize ACh again. Acetylcholine’s small amount diffuses outside synapses. It is also is being decomposed there via cholinesterase’s plasma, a soluble enzyme which can be found mainly in a blood serum as well as in liver cells (65).
Based on the acrylamide’s acute toxicity, research results in the animal models have concluded that regardless of the intake route, ACR caused neurotoxic symptoms. Above-mentioned symptoms consisted of coordination abnormalities as well as back legs weakening and paralysis (72-74). The histopathologic research has mainly shown axons and Schwann’s cells degeneration in peripheral nerves as well as in the spinal cord. The NOEL’s acute neurotoxicity value for rats was establish around the level of 0.5 mg/kg b.w./per day (46, 75). Johnson’s investigations (76), in which rats were fed with F 344 for two years and with acrylamide in water demonstrated acrylamide is a carcinogenic substance. The raise in the number of thyroids, nuclei, adrenal cancers in rats was demonstrated after the administration of 0.5 mg/kg dose per b.w. per day (77).
Kopanska et al. (78, 79) demonstrated some significant changes in ACh’s activity in mice brain and the muscles. It appears that AChE’s activity, which is an enzyme connected to cholinergic transformation, may be an important determinant of ACR’s neurotoxic properties (Fig. 3).
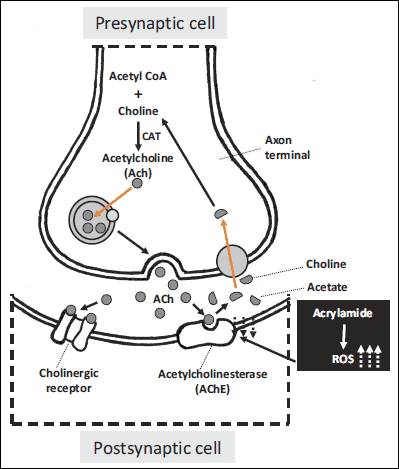 |
Fig. 3. The effect of acrylamide on the cholinergic synapse function (78, 79). |
Cholinergic nerves are involved in many of the brain’s functions among others memory, emotions stress reactions, a control of movement system as well as of basic organism’s functions. Cholinergic synapses are located in all preganglionic automotive fibers, also in some parasympathetic fibers that lie behind (69). The autonomic nervous system consists of nerves, ganglia and plexus which innervate heart, blood vessels, glands and also fibroids in different tissues. The sympathetic nervous system, due to its functions, increases the heart rate and the blood pressure switching the blood flow from visceral area and skin to skeletal muscles whereas the parasympathetic system reduces the heart rate, lowers the blood pressure and among others affects small intestine’s muscles and facilitates the absorption of nutrients (80). Cholinergic nerve functions are closely related with two chemical compounds, which are ACh and AChE. Acetylcholinesterase’s activity is adjusted to the level of the synaptic gap released ACh. Studies have shown that changes in an AChE’s activity influenced a rate of an ACh’s hydrolise and thus modifies a cholinergic synapses conduction (81). It has been demonstrated that ACR’s blocks an axonal transport through microtubule’s binding (30). It has recently been demonstrated that ACR’s shows a neurotoxic activity through a binding with rich in cysteine receptor’s proteins which in turn take part in a presynaptic release of a neurotransmitter (a component sensitive to N-ethylmaleimide), membranous reuptake (membranous dopamine’s transporter) and vesicular neuroconduction (vesicular moanine’s transporter). Additionally, it has been shown that dopamine levels increase significantly in response to ACR’s intoxication in rat striatum (82). It causes a disturbance of neuroconductions and nerve function which ultimately leads to morphological changes such as for example axons swelling and biochemical changes such as a Na+/K+-ATPase’s activity disturbance or visible neurotoxicity (28, 83). There is ample evidence of acrylamide acting directly on peripheral nerves and causing structural damage and functional changes. The ultrastructure’s damage (e.g. formation of grave disturbances in the function of a cell membrane) and functional changes (e.g. reduced release of neurotransmitters) suggest acrylamide distorts a fusion of synaptic vesicles (29, 84). The link between the presynaptic membrane and the synaptic vesicle is a foundation of neurophysiological processes and is key inthe process of building axonal endings and neurotransmitters releasing. Acrylamide disrupts the connections between membranes and hence has a toxic influence on the fusion of synaptic vesicles and the membrane of a presynaptic element, it also causes a degeneration of peripheral nerves thus reducing a neurotransmission (30).
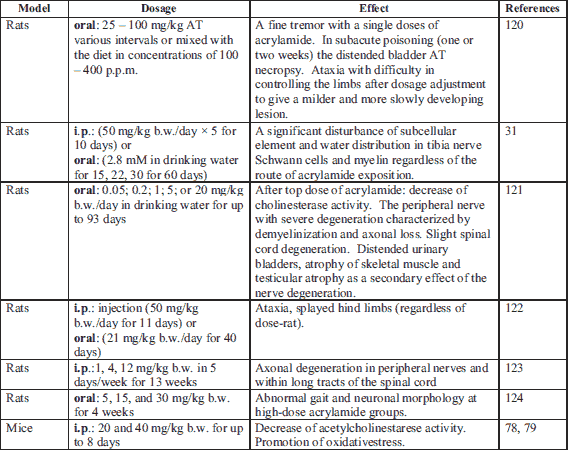
Many factors which might influence an AChE’s activity have been discovered so far. They are, among other, skainic acid, alcohol, pesticides, many drugs and free radicals (81, 85, 86). The characteristic of ACR’s exposure is an axon’s degeneration in the central and peripheral nervous system (87). The peripheral neuropathy and the creation of the hemoglobin’s addictive are symptoms which were observed both in humans who had occupational ACR contact, and in lab animals which were fed intraperitoneally or via ingestion (88). Kopanska et al. (78, 79) research shows ACR as a toxin has a significant influence on mice brain and muscle AChE’s activity. This compound blocked AChE’s activity. Data on ACR’s influence on AChE’s activity in cholinergic neurons are few and inconclusive in spite of some intensive research related to an ACR’s neurotoxicity. Pennisi et al. (47) have concluded ACR increases AChE’s activity in peripheral cholinergic neurons. Other studies have demonstrated that people who had been exposed to ACR, showed symptoms of neuropathy in the peripheral nerves. Symptoms of these were: muscle weakness, asthenia, fluctuating stimuli sensitivity or limb tingling. Rat’s four weeks ACR’s exposition caused a complete paralysis of the back limbs because of neurotransmission disturbance (87, 88). It seems there are a few mechanisms intermediating in an ACR’s influence on cholinergic synapses. Due to a biotransformation, ACR is partially oxidized to 2,3-epoxypropionamide which is also known as glycidamide. Both glycidamide and ACR constitute the DNA’s adducitives (89) and react with compounds which have-SH, -NH 2 or -OH groups in their structure (25). As a consequence, both substances take part in the modification of a genetic expression, mutation’s induction and in disturbances of cell’s energy transfers (90-94). One of the most significant mechanisms of ACR’s neurotoxic activity is the addictive forming out of the protein’s thiol groups, which play a decisive role in the neurotransmitter’s release regulation. Due to a docking, synaptic vesicles place themselves in the area of an active sphere. Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE),which belong to the trans membranes proteins family and which take part in recognizing and fusion of follicles and cell membranes, also take part in this mechanism (95). Synaptobrewin (v-SNARE), or a protein connected to a synaptic vesicle binds with presynaptic membranous protein, syntaxin (t-SNARE) which is closely connected to a voltage related calcium channel. The above-mentioned proteins provide an optimal location of the synaptic vesicle in relation to the place where the calcium signal occurs. Both of these proteins, in connection with a third SNAP-25 protein, which mainly takes part in the docking process, are targeted by toxins which are strong inhibitors of neurotransmitter’s release processes (96-98). The described mechanism covers both a neurotransmitter’s release to a synaptic gap and a presynaptic capture of this neurotransmitter (33). Defects related to an ACR’s presence in the central and peripheral nervous system, therefore might be caused by a handicapof neurotransmission (33). The following research confirms ACR reacts with cysteine’s residuum which can be found in presynaptic membranes of proteins. Its toxicity depends on a given protein’s role in the neurotransmission process (33) (Table 3).
i.p., intraperitoneal injection.
Brain and peripheral structures are particularly exposed to an oxidative stress (43). The consequence of an oxidative stress with flawed repair mechanisms of a cell is cell membranes oxidation, a change in structures and thus the protein’s function modification, which in turn leads to DNA damage (99, 100). The damage of enzymatic protein’s particles is a consequence of an activity of reactive oxygen forms. It leads to both structural and biological activity changes. The oxidative modification of proteins depends both on the amino acid’s residues and a prosthetic group modification (101). The entire protein parts get fragmentarised as well (101). There is data that exist about the influence of free radicals on the blocking of an AChE’s activity. This effect was observed both in the brain and in peripheral cholinergic neurons (102, 103). The oxidative stress is a very important factor in a pathogenesis ofthe nervous system’s neurodegenerative diseases. Both ACR and glicydamide raise a production of oxygen’s reactive forms and disturb a nervous system’s redox balance (41). ROS are extremely toxic to nerve cells due to their activity on the neurolema’s polyunsaturated fatty acids (104). The free radical’s attack causes a decline in the neurolemma’s liquidity which also leads to damage in the insides of the nerve cells, which can be seen as lipids and proteins oxidation, a damage to both the cell nucleus and mitochondria, Na/K-ATP-ase’s inhibition, ion’s transport impairment (105). The disturbance of cholinergic neurons activity is observed in neurodegenerative maladies such as Alzheimer’s and Parkinson’s diseases. The direct cause of an Alzheimer’s disease is two amyloid β-proteins deposition in the brain which builds up in diffusion and neurotic plates and MAP as a form of neurofilaments. There is a permanent and progressive damage of the cholinergic system and a continuous decline in an acetylcholine release (61, 66). The cholinergic system plays a vital role in learning and memory, where Alzheimer’s disease is directly connected with a disfunction of the cholinergic system (11). Acetylcholinesterase exhibits a peptidase’s activity which is extremely important in Alzheimer’s pathogenesis. Thanks to a peptidase’s activity, AChE breaks down an amyloid’s precursor protein in a place which is not amyloidogenic (12-14). There is a similar phenomenon observed in the case of elderly people (69). It has been recently suggested that the pure protein of AChE’s enzyme may induce neurological changes in Alzheimer’s disease. It has been concluded AChE β-amyloid complex is much more toxic in comparison with amyloid and it causes greater neurological changes while a pure AChE causes the creation of amyloid due to the heightened expression of the protein’s precursor in glial cells (15). AChE also plays a direct role in the causing of apoptosis in nerve cells (62). The acetylcholinesterase’s activity disturbances may have a direct connection with Parkinson’s pathogenesis for there is not only an AChE’s activity decline observed in 30% of the sick population, but also some quantitative changes of certain particle forms are being observed. In connection with the above-mentioned facts, it seems accurate to state that acrylamide may be a significant factor in a pathogenesis of neurodegenerative diseases.
Concluding remarks
Many researchers have extensively studied the various aspects of neurotoxic effects on the central and peripheral nervous system. Wyrembek et al. (106) have investigated that the neurodevelopmental disorders in children arising from different microelements such as mercurials evoking electrophysiological changes in excitatory and inhibitory neurons and neurotransmitters including GABA and NMDA receptors as determined in cultured neurons with the mode of neuronal action. In another interesting research paper about the ACR toxicity, guinea pigs were treated prenatally with ACR which affected the intestinal structure elements and perhaps, would influence the physiology of intestinal digestion in later ages of these animals (107).
In 2002, SFA concluded, there are products with an exceptionally high ACR’s content. It was a starting point towards new research into an above-mentioned substance. This is a very well-known fact that the group of ACR products consists of among others those products which were heat treated (108). ACR’s average consumption in adults equals 0.5 mg/kg of body weight (109-111). Children’s average consumption equals roughly around 0.6 mg/kg. This stems from the fact that children have lower body weight, and therefore they intake a greater ACR’s dose calculated on 1 kg/b.w. Furthermore, there are products ‘rich’ in the substance such as French fries and potato chips which are eaten more often in this younger age group among others. A survey results by Larsson et al. (112) estimated that an average daily adult’s consumption of acrylamide equals 36.1 µg, whereas the main acrylamide's sources were coffee (23%), whole wheat bread (17%), crispbread (8%), white bread (7%), cakes/buns (7%), waffels/crackers/rusks (6%), cereal (6%), fried potatoes (6%).
It is also important that exposure to acrylamide is not only via diet. People working on the construction sites where ACR and N-methylacrylamide’s derivative is used (112) or people employed to produce polyacrylamide as well as people working in biomedical labs are also being exposed to the compound’s toxicity (114, 115). Occupational hazard may lead to symptoms of a peripheral neuropathy which is evidence of ACR’s high dose intake (84). Based on studies which were carried out on lab animals, it has been concluded ACR damages cells both in the nervous and reproductive systems, furthermore it contributes to the occurrence of cancer in hormone-dependent tissues (116, 117). Research results point to neurotoxic (32, 118), genotoxic (119, 120) and carcinogenic (121-123) ACR’s influences, regardless of the way it enters a human organism. Until now, we focused on the effect of ACR on the cholinergic system. In the nearest future, we want to expand our research and open a unique approach to studying the interaction between toxic effects of ACR on blood-brain barrier modification.
Conflict of interests: None declared.
REFERENCES
- Ericsson S. Acrylamide in Food Products: Identification, Formation and Analytical Methodology. Institutionen for Miljokemi, Stockholms Universitet 2005, pp. 9-53.
- Girma KB, Lorenz V, Blaurock S, Edelmann F. Coordination chemistry of acrylamide. Coord Chem Rev 2005; 249: 1283-1293.
- Zyzelewicz D, Nebesny E, Oracz J. Acrylamide - formation, physicochemical and biological properties [in Polish]. Bromatol Chem Toksykol 2010; 43: 415-427.
- Friedman M. Chemistry, biochemistry, and safety of acrylamide. Rev J Agric Food Chem 2003; 51: 4504-4526.
- Becalski A, Brady B, Feng S, Gauthier BR, Zhao T. Formation of acrylamide at temperatures lower than 100°C: the case of prunes and a model study. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 2011; 28: 726-730.
- Szumska M, Janoszak B, Bodzek D. Acrylamide. Mechanism of formation and occurrence in food. Part 1 [in Polish]. Bromatol Chem Toksykol 2003; 36: 347-354.
- Jagerstad M, Skog K. Genotoxicity of heat-processed foods. Mutat Res 2005; 574: 156-172.
- Chico Galdo V, Massart C, Jin L, et al. Acrylamide anin vitro thyroid carcinogenic agent, induces DNA damage in rat thyroid cell lines and primary cultures. Mol Cell Endocrinol 2006; 257-258: 6-14.
- Mustafa A, Kamal-Eldin A, Petersson EV, Andersson R, Aman P. Effect of acrylamide content in fresh and stored rye crisp bread. J Food Compost Anal 2008; 21: 351-355.
- Rice JM. The carcinogenicity of acrylamide. Mutat Res 2005; 580: 3-20.
- Yamada S, Nebeshima T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol Ther 2000; 88; 93-113.
- Dave KR, Syal AR, Katyare SS. Tissue cholinesterase. A comparative study of their kinetic properties. Z Naturforsch C 2000; 55: 100-108.
- Balasubramanian AS. Amyloid beta peptide processing, insulin degrading enzyme and butyrylcholinesterase. Neurochem Res 2001; 26: 453-456.
- Darvesh S, Martin E, Walsh R, Rockwood K. Differential effects of lipid-lowering agents on human cholinesterases. Clin Biochem 2004; 37: 42-49.
- Beckman M. Untangling Alzheimer’s by paring plaques bolsters amyloid theory. Science 2004; 305: 762.
- Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption. Official Journal of the European Communities 1998; 41 (L330): 32-54.
- Twenty-sixth Commission Directive 2002/34/EC of 15 April 2002 adapting to technical progress Annexes II, III and VII to Council Directive 76/768/EEC on the Approximation of the Laws of the Member States Relating to Cosmetic Products. Official Journal of the European Communities 2002; 45 (L102): 19-31.
- Klauing JE. Acrylamide carcinogenicity. J Agric Food Chem 2008; 56: 5984-5988.
- Besaratinia A, Pfeifer GP. DNA adduction and mutagenic properties of acrylamide. Mutat Res 2005; 580: 31-40.
- Sen A, Ozgun O, Arinc E, Arslan S. Diverse action of acrylamide on cytochrome P450 and glutathione S-transferase isozyme activities, mRNA levels and protein levels in human hepatocarcinoma cells. Cell Biol Toxicol 2012; 28: 175-186.
- Stadler RH, Scholz G. Acrylamide: an update on current knowledge in analysis, levels in food, mechanisms of formation, and potential strategies of control. Nutr Rev 2004; 62: 449-467.
- Szczerbina T, Banach Z, Tylko G, Pyza E. Toxic effects of acrylamide on survival, development and haemocytes of Musca domestica. Food Chem Toxicol 2008; 46: 2316-2319.
- Arslan S, Ozgun O, Celik G, Semiz A, Dusen O, Mammadov R. Effects of Cyclamen trochopteranthum on hepatic drug metabolizing enzymes. Arch Biol Sci Belgr 2011; 63: 545-555.
- Sorgel F, Weissenbacher R, Kinig-Schippers M, Hofmann A, Illauer M, Skott A. Acrylamide: increased concentrations in homemade food and first evidence of its variable absorption from food, variable metabolism and placental and breast milk transfer in humans. Chemotherapy 2002; 48: 267-274.
- Pingot D, Pyrzanowski K, Michalowicz J, Bukowska B. Toxicity of acrylamide and its metabolite-glicydamide [in Polish]. Med Pr 2013; 64: 259-271.
- Atay Z, Calgan D, Ozakat E, Varnail T. Acrylamide and glycidamide adducts of guanine. J Mol Struct 2005; 728: 249-251.
- Hagmar L, Tornqvist M, Nordander C, et al. Effects of occupational exposure to acrylamide using hemoglobin adducts as biomarkers of internal dose. Scand J Work Environ Health 2001; 27: 219-226.
- Yousef MI, El-Demerdash FM. Acrylamide-induced oxidative stress and biochemical perturbations in rats. Toxicology 2006; 219: 133-141.
- Fullerton PM. Electrophysiological and histological observations on peripheral nerves in acrylamide poisoning in man. J Neurol Neurosurg Psychiatry 1969; 32: 186-192.
- LoPachin RM. The changing view of acrylamide neurotoxicity. Neurotoxicology 2004; 25: 617-630.
- LoPachin RM, Castiglia CM, Saubermann AJ. ACR disrupts elemental composition and water content of rat tibial nerve. I. Myelinated axons. Toxicol Appl Pharmacol 1992; 115: 21-22.
- Sheng Q, Zou H, Lu Z, et al. Effects of acrylamide on the activity and structure of human brain creatine kinase. Int. J Mol Sci 2009; 10: 4210-4222.
- Barber DS, Hunt JR, Ehrich MF, Lehning EJ, LoPachin RM. Metabolism, toxicokinetics and hemoglobin adduct formation in rats following subacute and subchronic acrylamide dosing. Neurotoxicology 2001; 22: 341-353.
- The Advisory Committee on Safety and Health at Work. Opinion on the Approach and Content of an Envisaged Proposal by the Commission on the Amendment of Directive 2004/37/EC on Carcinogens and Mutagens at the Workplace. Doc. 727/13. 2012 Adopted on 30/05/2013, https://www.etui.org
- Batoryna M, Lis MW, Formicki G. Antioxidant defence in the brain of 1-d-old chickens exposed in ovo to acrylamide. Br Poult Sci 2018; 59: 198-204.
- Erdemli ME, Aksungur Z, Gul M, et al. The effects of acrylamide and vitamin E on kidneys in pregnancy: an experimental study. J Matern Fetal Neonatal Med 2018; 59: 198-204.
- Schlesier K, Harwat M, Bohm V, et al. Assessment of antioxidant activity by using differentin vitro methods. Free Radic Res 2002; 36: 177-187.
- Meister A. Selective modifications of glutathione metabolism. Science 1984; 220: 472-477.
- Itoh K, Ishii T, Wakabayashi N, Yamamoto M. Regulatory mechanism of cellular response to oxidative stress. Free Radic Res 1999; 31: 319-325.
- Cooper AJ, Kristal BS. Multiple roles of glutathione in the central nervous system. Biol Chem 1997; 378: 793802 .
- Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med 2002; 33: 37-44.
- Vertuani S, Angusti A, Manfredini S. The antioxidants and pro-antioxidants network: an overview. Curr Pharm Des 2004; 14: 1677-1694.
- Acaroz U, Ince S, Arslan-Acatoz D, et al. The ameliorative effects of boron against acrylamide-induced oxidative stress, inflammatory response, and metabolic changes in rats. Food Chem Toxicol 2018; 118: 745-752.
- Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med 2002; 227: 671-682.
- Choi J, Lee KJ, Zheng Y, Yamaga AK, Lai MM, Ou JH. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 2004; 39: 81-89.
- Klaunig JE, Kamendulis LM. Mechanism of acrylamide induced rodent carcinogenicity. In: Chemistry and Safety of Acrylamide in Food. Springer Science, Inc. 2005.
- Pennisi M, Malaguarnera G, Puglisi V, Vinciguerra L, Vacante M, Malaguarnera M. Neurotoxicity of acrylamide in exposed workers. Int J Environ Res Public Health 2013; 10: 3843-3854.
- Zhu Y, Zeng T, Yu S, et al. Effects of acrylamide on the nervous tissue antioxidant system and sciatic nerve electrophysiology in the rat. Neurochem Res 2008; 33: 2310-2317.
- Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 1995; 38: 22-33.
- Dean B, McLeod M, Keriakous D, Mckenzie J, Scarr E. Decreased muscarinic receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psych 2002; 7: 1083-1091.
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer’s disease and the basal forebrain cholinergic system: relation to beta-amyloid peptides, cognition and treatment strategies. Prog Neurobiol 2002; 68: 209-245.
- Ma L, Seager MA, Wittman M, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA 2009; 106: 15950-15955.
- Kitamura N, Araya R, Kudoh M, et al. Beneficial effects of estrogen in a mouse model of cerebrovascular insufficiency. PLoS One 2009; 4(4): e5159. doi: 10.1371/ journal.pone.0005159
- Dani JA. Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry 2001; 49: 166-174.
- Luo W, Latchney LR, Culp DJ. D protein coupling to M1 and M3 muscarinic receptors in subligual glands. Am J Physiol Cell Physiol 2001; 280: 884-896.
- Bymaster FP, McKinzie DL, Felder CC, Wess J. Use of M1-M5 muscarinic receptor knockout mice as a novel tools to declinate the physiological roles of the muscarinic cholinergic system. Neurochem Res 2003; 28: 437-442.
- Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol 2000; 61: 75-111.
- Messer WS. The utility of muscarninic agonists in the treatment of Alheimer’s disease. J Mol Neurosci 2002; 19: 187-193.
- Dean B. Evolution of the human CNS cholinergic system. Has this resulted in the emrgence of psychiatric disease? Aust NZ J Psychiatry 2009; 43: 1016-1028.
- Tougu V. Acetylocholinoesterase: mechanism of catalysis and inhibition. Curr Med Chem Cent Nerv Syst Agents 2001; 1: 155-170.
- Giacobini E. Cholinesterase inhibitors: from the calabar bean to Alzheimer therapy. In: Cholinesterases and Cholinesterase Inhibitors: Basic Preclinical and Clinical Aspects (Giacobini E, ed.). CRC Press, 2000, 181-226.
- Stephanson J, Czepulkowski B, Hirst W, Mufti G. Deletion of the acetylcholinesterase locus at 7q22 associated with meylodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leuk Res 1996; 20: 235-241.
- Knipper M, Khale C, Breer H. Purification and reconstruction of the high affinity acetylcholine transporter. Biochem Biophys Acta 1991; 1065: 107-113.
- Cavdar H, Senturk M, Guney M, el al. Inhibition of acetylcholinesterase and butyrylcholinesterase with uracil derivatives: kinetic and computational studies. J Enzyme Inhib Med Chem 2019; 34: 429-437.
- Barbosa M, Rios O, Velasquez M, Villalobos J, Ehrmanns J. Acetylcholinesterase and butyrylcholinesterase histochemical activities and tumor cell growth in several brain tumors. Surg Neurol 2001; 55: 106-112.
- Li B, Stribley JA, Ticu A, Xie W, Schopfer LM, Hammond P. Abundant tissue butyrylcholin-esterase and ist possible function in the acetylcholin-esterase knockout mouse. J Neurochem 2012; 75: 1320-1331.
- Wonnacott S. Presynaptic nicotinic acetylocholine receptors. Trends Neurosci 1997; 20: 90-105.
- Massoulie J, Pezzementi L, Bon S, Krejci E, Valette FM. Molecular and cellular biology of cholinesterasses. Prog Neurobiol 1993; 41: 31-91.
- Meslam M, Guillozet A, Shaw P, Quinn B. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol Dis 2002; 9: 88-93.
- Yamada M, Ichinose M. The cholinergic anti-inflammatory pathway: an innovative treatment strategy for respiratory diseases and their comorbidities. Curr Opin Pharmacol 2018; 40: 18-25.
- Lee HW, Pyo S. Acrylamide induces adipocyte differentiation and obesity in mice. Chem Biol Interact 2019; 298: 24-34.
- Freisling H. Dietary acrylamide intake of adults in the European Prospective Investigation into Cancer and Nutrition differs greatly according to geographical region. Eur J Nutr 2013; 52: 1369-1380.
- Hogervorst JG. The carcinogenicity of dietary acrylamide intake: a comparative discussion of epidemiological and experimental animal research. Crit Rev Toxicol 2010; 40: 485-512.
- Li J, Li D, Yang Y, Xu T, Li P, He D. Acrylamide induces locomotor defects and degeneration of dopamine neurons in Caenorhabditis elegans. J Appl Toxicol 2016; 36: 60-67.
- Manjanatha MG, Aidoo A, Shelton SD, et al. Genotoxicity of acrylamide and its metabolite glycidamide administered in drinking water to male and female Big Blue mice. Environ Mol Mutagen 2006; 47: 6-17.
- Johnson KA. Chronic toxicity and oncogenicity study on acrylamide incorporated in the drinking water of Fischer 344 rats. Toxicol Appl Pharmacol 1986; 85: 154-168.
- Ruden C. Acrylamide and cancer risk – expert risk assessments and the public debate. Food Chem Toxicol 2004; 42: 335-349.
- Kopanska M, Lukac N, Kapusta E, Formicki G. Acrylamide influence on activity of acetylcholinesterase, thiol groups, and malondialdehyde content in the brain of Swiss mice. J Biochem Mol Toxicol 2015; 29: 472-478.
- Kopanska M, Czech J, Zagata P, Dobrek L, Thor P, Formicki G. Effect of the different doses of acrylamide on acetylocholinoesterase activity, thiol groups, malondialdehyde concentrations in hypothalamus and selected muscles of mice. J Physiol Pharmacol 2017; 68: 565-571.
- Rosenberry TL. New inhibitors of the peripheral site in acetylocholinestarase that specifically block organophosphotylation. Technical Report. Mayo Clinic Jacksonville 2000. https://apps.dtic.mil/dtic/tr/fulltext/u2/ b259791.pdf
- Sahitya C, Rajbanshi S, Kunala P, Pandanaboina S, Gopalreddy V, Wudayagiri R. Tramadol safety - cholinergic system of rat brain without nociception. Acta Pol Pharm 2012; 69: 833-841.
- Pan X, Guo X, Xiong F, Cheng G, Lu Q, Yan H. Acrylamide increases dopamine levels by affecting dopamine transport and metabolism related genes in the striatal dopaminergic system. Toxicol Lett 2015; 236: 60-68.
- Ghorbel I, Amara IB, Ktari N, et al. Aluminium and acrylamide disrupt cerebellum redox states, cholinergic function and membrane-bound ATPase in adult rats and their offspring. Biol Trace Element Res 2016; 174: 335-346.
- Zhang B, Shao H, Wang XH, et al. Acrylamide-induced subacute neurotoxic effects on the cerebral cortex and cerebellum at the synapse level in rats. Biomed Environ Sci 2017; 6: 432-443.
- Szaroma W. The influence of kainic acid on acetylcholinesterase activity in the brain and muscles of the mouse. Part I. After blocking muscarinic receptors. Acta Biol Crac (Zool) 1997; 39: 63-69.
- Ehrich M, Van Tassell R, Li Y, Zhou Z, Kepley CL. Fullerene antioxidants decrease organophosphate-induced acetylcholinesterase inhibitionin vitro. Toxicolin vitro 2011; 25: 301-307.
- Odland L, Romert L, Clemedson C, Walum E. Glutathione content, glutathione transferase activity and lipid peroxidation in acrylamide-treated neuroblastoma N1E 115 cells. Toxicolin vitro 1994; 8: 263-267.
- Konings CH, Kuiper MA, Mulder C, Calliauw J, Wolters EC. CSF acetylcholinesterase in Parkinson disease: decreased enzyme activity and immunorectivity in demented patients. Clin Chim Acta 1995; 235: 101-105.
- Dybing E, Farmer PB. Human exposure and internal dose assessments of acrylamide in food. Food Chem Toxicol 2005; 43: 365-410.
- Dearfield KL, Douglas GR, Ehling UH, Moore MM, Sega GA, Brusick DJ. Acrylamide: a review of its genotoxicity and an assessment of heritable genetic risk. Mutat Res 1995; 330: 71-99.
- Koyama N, Sakamoto H, Sakuraba M, et al. Genotoxicity of acrylamide and glycidamide in human lymphoblastoid TK6 cells. Mutat Res 2006; 603: 151-158.
- Martins C, Oliveira NG, Pingarilho M, et al. Cytogenetic damage induced by acrylamide and glycidamide in mammalian cells: correlation with specific glycidamide DNA adducts. Toxicol Sci 2007; 95: 383-339.
- Mei N, Hu J, Churchwell MI, et al. Genotoxic effects of acrylamide and glycidamide in mouse lymphoma cells. Food Chem Toxicol 2008; 46: 628-663.
- Von Tunglen LS, Doerge DR, Gamboa da Costa G, et al. Tumorigenicity of acrylamide and its metabolite glycidamide in the neonatal mouse bioassay. Int J Cancer 2012; 131: 2008-2015.
- Lin RC, Scheller RH. Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 2000; 16: 19-49.
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature 1994; 368: 823-828.
- Nicholls DG. Proteins, Transmitters and Synapses. Oxford, Blackwell Scientific Publications, 1994.
- Revest P, Longstaff A. Molecular Neuroscience. Oxford, BIOS Scientific Publishers Ltd, 1998.
- Van Remmen H, Hamilton ML, Richardson A. Oxidative damage to DNA and aging. Exerc Sport Sci Rev 2003; 31: 149-153.
- Komoike Y, Matsuoka M.in vitro andin vivo studies of oxidative stress responses against acrylamide toxicity in zebrafish. J Hazard Mater 2019; 365: 430-439.
- Forsberg L, de Faire U, Morgenstern R. Effect of basal level of antioxidants on oxidative DNA damage in humans. Eur J Nutr 2007; 46: 174-181.
- Dos Santos AA, Dos Santos DB, Ribeiro RP, et al. Effects of K074 and pralidoxime on antioxidant and acetylcholinesterase response in malathion-poisoned mice. Neurotoxicology 2011; 32: 888-895.
- Kazi AI, Oommen A. Monocrotophos induced oxidative damage associates with severe acetylcholinesterase inhibition in rat brain. Neurotoxicology 2012; 33: 156-161.
- Venkataswamy M, Divya K, Pallavi C, Thyagraju K. Characterization of glutathione-s-transferases-suppression of antioxidant enzymes by acrylamide in developing chick embryonic brain. Int J Pharm Biol Sci 2013; 4: 668-677.
- Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci 1994; 4: 2924-2932.
- Wyrembek P, Szczuraszek K, Majewska MD, Mozrzymas JW. Intermingled modulatory and neurotoxic effects of thimerosal and mercuric ions on electrophysiological responses to GABA and NMDA in hippocampal neurons. J Physiol Pharmacol 2010; 61: 753-758.
- Tomaszewska E, Dobrowolski P, Puzio I, et al. Acrylamide-induced prenatal programming of intestine structure in guinea pig. J Physiol Pharmacol 2014; 65: 107-115.
- Baum M, Fauth E, Fritzen S, et al. Acrylamide and glicidamide: approach towards risk assessment based on biomarker-guided dosimetry of genotoxic/mutagenic effects in human blood. Adv Exp Med Biol 2005; 561: 77-88.
- Claus A, Carle R, Schieber A. Acrylamide in cereal products: a review. J Cereal Sci 2008; 47: 118-113.
- Mucci LA, Wilson KM. Acrylamide intake through diet and human cancer risk. J Agric Food Chem 2008; 56: 6013-6019.
- Pelucchi C, La Vecchia C, Bosetti C, Boyle P, Boffetta P. Exposure to acrylamide and human cancer – a review and meta-analysis of epidemiologic studies. Ann Oncol 2011; 22: 1487-1499.
- Larsson CS, Akesson A, Bergkvist L, Wolk A. Dietary acrylamide intake and risk of colorectal cancer in a prospective cohort of men. Eur J Cancer 2009; 45: 513-516.
- Bergmark E, Calleman CJ, He F, Costa LG. Determination of hemoglobin adducts in humans occupationally exposed to acrylamide. Toxicol Appl Pharmacol 1993; 120: 45-54.
- Pantusa VP, Stock TH, Morandi MT, Harrist RB, Afshar M. Inhalation exposures to acrylamide in biomedical laboratories. AIHA J (Fairfax, Va) 2002; 63: 468-470.
- Katen AL, Roman D. The genetic consequences of paternal acrylamide exposure and potential for amelioration. Mutat Res 2015; 777: 91-100.
- Chen JH, Chou CC. Acrylamide inhibits cellular differentiation of human neuroblastoma and glioblastoma cells. Food Chem Toxicol 2015; 82: 27-35.
- LoPachin RM, Barber DS. Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Toxicol Sci 2006; 94: 240-255.
- Gamboa da Costa G, Churchwell MI, Hamilton LP, et al. DNA adduct formation from acrylamide via conversion to glycidamide in adult and neonatal mice. Chem Res Toxicol 2003; 16: 1328-1337.
- Blasiak J, Gloc E, Wozniak K, Czechowska A. Genotoxicity of acrylamide in humans lymphocytes. Chem Biol Interact 2004; 149: 137-149.
- Fullerton PM, Barnes, JM. Peripheral neuropathy in rats produced by acrylamide. Br J Ind Med 1966; 23: 210-221.
- Burek JD, Albee RR, Beyer JE, et al. Subchronic toxicity of acrylamide administered to rats in the drinking water followed by up to 144 days of recovery J Environ Pathol Toxicol 1980; 4: 157-182.
- LoPachin RM, Ross JF, Reid ML, Dasgupta S, Mansukhani S, Lehning EJ. Neurological evaluation of toxic axonopathies in rats: acrylamide and 2,5-hexanedione. Neurotoxicology 2002; 23: 95-110.
- Moser VC, Anthony DC, Sette WF. Comparison of subchronic neurotoxicity of 2-hydroxyethyl acrylate and acrylamide in rats. Appl Toxicol 1992; 18: 343-352.
- Tian SM, Ma YX, Shi J, Lou TY, Liu SS, Li GY. Acrylamide neurotoxicity on the cerebrum of weaning rats. Neural Regen Res 2015; 10: 938-943.
A c c e p t e d : December 30, 2018