VULNERABILITY OF GASTRIC AND SMALL INTESTINAL MUCOSA TO ULCEROGENIC ACTION OF INDOMETHACIN IN C57/BL6/J MICE AND TRANSIENT RECEPTOR POTENTIAL CHANNEL VANILLOID TYPE 1 KNOCKOUT MICE
INTRODUCTION
Capsaicin is a compound found in chili peppers and responsible for their burning and irritant effect. Capsaicin demonstrates a wide variety of biological effects that make it the target of extensive researches (1-3). As a member of the vanilloid family, it binds to a vanilloid receptor type 1 (TRPV1, transient receptor potential vanilloid 1), named also capsaicin receptor. TRPV1 receptor, a nonselective cation channel with six transmembrane domains, is activated by a variety of stimuli including capsaicin and endogenous vanilloids as well as extracellular protons, noxious temperature (>43°C) and some lipid derivatives (4). TRPV1 receptors are expressed selectively in the cell membrane of C and Ad afferent fibers that is sensitized by noxious heat and inflammatory mediators. TRPV1 receptors are involved in processing of pain stimuli and inflammation (5, 6). TRPV1 receptor is considered as an essential component of the cellular signaling mechanism through which injury produces thermal hyperalgesia and pain hypersensitivity. Genetically engineered knockout mice lacking TRPV1 receptor show partial deficits in acute thermal nociception supporting the role of this channel in heat-evoked pain (5).
Activation of TRPV1 receptors is accompanied by a release of calcitonin gene-related peptide (CGRP) and substance P (SP) that induce neurogenic inflammation characterized by vasodilatation, plasma extravasation and leukocyte migration (7-9). At the same time TRPV1 receptors may promote the release of substances that prevent the development of inflammation, such as somatostatin which may reach the circulation and elicit systemic anti-inflammatory and antihyperalgesic effect (10, 11).
In the gastrointestinal tract TRPV1 receptors are highly expressed by extrinsic sensory neurons and by intrinsic enteric neurons within the myenteric ganglia and interganglionic fibers (12-14). Their role in visceral hypersensitivity induced by inflammation in gastrointestinal tract is well documented (15-17). However, the involvement of TRPV1 receptors in the maintenance of the gastrointestinal mucosal integrity or/and injury remains obscure.
It is known that administration of capsaicin, agonist of TRPV1 receptors, in low doses reduces or prevents gastric mucosal injury induced by different irritants (hydrochloric acid, ammonia, ethanol, aspirin or indomethacin) (18). The gastroprotective action of capsaicin is abolished by repeated or prolonged capsaicin administration (19-21). Taken together the data indicate that TRPV1 receptors are involved in gastroprotection. However, pretreatment with TRPV1 receptor antagonist capsazepine also markedly reduces ethanol-induced lesions in rats suggesting that in this situation TRPV1 stimulation may initiate an adverse events that lead to gastric injury (8).
Little is known on the role of TRPV1 receptors under circumstances of the small intestinal injury. It is known that capsaicin as well as endogenous agonists of TRPV1 receptors anandamide and 2-arachidonoylglycerol cause inflammation in the rat ileum whereas capsazepine inhibits these effects (22, 23). The data suggest the pathological effect of TRPV1 receptor stimulation on the small intestinal mucosa.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used in clinic as analgesic and anti-inflammatory drugs; however they exert severe adverse effects on the gastrointestinal tract (24). Patients taking NSAIDs have an increased risk of ulceration not only in the stomach but also in the small intestine (24-26). Experimental animal studies with NSAIDs confirm that these drugs including indomethacin (IM) cause damage in the stomach as well as the small intestine (27-29).
Although indomethacin model of ulceration is extensively used in animal experiments functional role of TRPV1 receptors under IM-induced pathology in the stomach and the small intestine has not been directly studied. There are indirect data on the gastroprotective effect of capsaicin as agonist of TRPV1 receptors on IM-induced injury (18, 30). Our previous data (31) and data of literature (32) demonstrate that desensitization of capsaicin sensitive neurons aggravates IM-induced gastric erosions. Based on these data it may be assumed that TRPV1 receptors are involved in maintenance of gastric mucosa integrity under IM-induced injury. However, there are no data on the role of TRPV1 receptors under IM-induced injury in the small intestine.
One of common approaches to specifically evaluate the functional role of TRPV1 receptors is using TRPV1 gene-deficient mice (33). IM action on chronic pain under inflammatory conditions in TRPV1 knockout (TRPV1 KO) mice is well documented (34). Daily treatment of IM (1 mg/kg intraperitoneally) diminished arthritis and related hyperalgesia in both of TRPV1 KO and wild type mice but IM effects were lesser in TRPV1 KO mice (34). Nevertheless, IM action on the gastrointestinal tract in TRPV1 KO mice remains unknown.
It should be emphasized that the pathogenic mechanisms of NSAID-induced damage in the upper and the lower gastrointestinal tract have been studied separately (29, 35-37). The experimental designs to cause IM-induced damage in the stomach and the small intestine are also different. However, previously we developed experimental model allowing us to cause both gastric and small intestinal injury after single IM injection. We demonstrated that a single IM injection to pre-starving rats at a dose 35 mg/kg (the gastric ulcerogenic dose) may cause damage in the stomach and then, after refeeding, in the small intestine (38, 39). Thus, the pathological process is consistently spreading from the stomach to the small intestine: healing of indomethacin-induced gastric erosions is followed by an appearance of injury in the small intestine. It means that IM-induced injury in the stomach and the small intestine should not be considered separately. We used here this approach (single IM administration) for studying of a functional role of TRPV1 receptors under circumstances of IM-induced pathological process in the stomach and the small intestine.
We showed previously that glucocorticoids released in response to acute stress or NSAIDs are naturally occurring protective factors that play an important role in maintenance of the gastric mucosal integrity. They exert gastroprotective actions in co-operation with prostaglandins (PGs), NO, and capsaicin-sensitive afferent neurons; and their compensatory gastroprotective action is observed when the protective mechanism provided by either of these factors is impaired (31). Nevertheless, contribution of glucocorticoids in gastroprotective mechanisms in TRPV1 gene-deficient mice remains unclear.
The present study was performed to investigate and compare the vulnerability of gastric as well as small intestinal mucosa to ulcerogenic action of indomethacin in mice with genetically deleted TRPV1 receptor (TRPV1 KO) and in C57/BL6/J mice. Because compensatory gastroprotective role of glucocorticoids during inhibition of PGs production and desensitization of capsaicin-sensitive sensory neurons has been shown previously (31) we also compared plasma corticosterone levels C57BL6/J mice and TRPV1 KO mice. Taking into consideration a crucial role of TRPV1 receptors in heat evoked pain (5, 6) we tested somatic pain sensitivity in C57BL6/J mice and TRPV1 KO mice under thermal stimulus using tail flick test.
MATERIALS AND METHODS
Animals
Experiments were performed on male mice weighing 30 – 40 g. C57/BL6/J mice as well as TRPV1 receptor gene knockout mice, which are descended from parent groups purchased in the Jackson laboratory (Bar Harbor, ME), were used in experiments.
All mice (keeping 4 – 5 animals per cage) were maintained under an artificial 12 h dark-light cycle (lights on at 8:00 am) at a temperature of 22 – 24°C. They were provided with the laboratory chow and water ad libitum.
Care and treatment of animals were according to the European Communities Council guidelines on animal research (86/609/EEC) and the local animal care committee at the Pavlov Institute of Physiology Russian Academy of Sciences.
Drugs
We used indomethacin (Sigma, Germany) for induction of gastrointestinal injury. Indomethacin suspension was prepared immediately before administration using physiological saline supplemented with drops of Tween 80 (Sigma, Germany) as vehicle.
Induction and assessment of the gastric and the small intestine injury
To induce gastrointestinal injury IM was injected into pre-starving (24 h) mice. Access to food (refeeding) was restored 4 hours after IM administration. IM was injected subcutaneously in a single ulcerogenic dose (35 mg/kg in a volume of 10 ml/kg) at 11 o’clock. Control animals were given vehicle.
According to our previous data (38) gastric erosions developed 4 hours after single injection of IM at the dose of 35 mg/kg, then, they were healed for 48 hours. The healing of gastric erosions was followed by the small intestine injury which was visible 24, 48 or 72 hours after the IM injection. Based on these data, here we examined IM-induced gastric injury 4 after IM administration and small intestinal injury 24, 48 and 72 hours after IM injection.
The total areas (mm2) of the gastric and small intestinal damage were estimated by using the image analysis software (Image J). The total area of the gastric injury for each mouse included the areas of all the hemorrhagic erosions within glandular part of stomach; the total area of small intestinal injury for each mouse included macroscopically visible damages, hemorrhages and inflammatory areas within the small intestine. Dead animals were excluded from analysis.
In addition to macroscopic evaluation we performed a histological analysis of small intestinal injury. Small intestines were fixed in 10% neutral buffered formalin and embedded into paraffin after dehydration in a series of isopropyl alcohols. Then, the 4 µm sections were stained using Mayer’s hematoxylin and eosin (Emmonia Biothech Chelopech, Bulgaria). Injury was evaluated at 100 fold magnification in 15 – 20 fields of view in the small intestine. The histological assessment of damage was made using a five-score scale included the following parameters (40): intestinal villi without visible (or with small) changes like intact state (1); partial detachment of enterocyte epithelium, shortening of villi (2); weak (partial) destruction of intestinal villi as well as leukocytes infiltration of lamina propria (3); severe (intensive necrosis) destruction of intestinal villi as well as leukocytes infiltration of lamina propria and severe denudation of epithelium (4); full destruction of intestinal villi (their residues are empty) (5). The injury was estimated blindly.
Nociceptive testing
Somatic pain sensitivity was tested by tail flick latency (tail flick test) using tail-flick meter (Panlab, Harvard Apparatus, Spain). Mice were placed into the container to restrict softly their movements and, then, allowed them to habituate for 2 minutes. Tail flick reflex was induced by tail thermal stimulation which was discontinued after 10 seconds to avoid tissue damages (the cutoff point was 10 s), the focus (decimal selector of heat intensity) was set up 40. Tail flick latency was measured as the time from the onset of stimulus exposure till tail flick reflex and was automathically registered by computer program. For each animal, the tail flick latency was obtained as the mean of three measurements.
Collection of blood samples and estimation of plasma corticosterone levels
Immediately after nociceptive testing mice were decapitated and trunk blood was collected. Blood was transferred to micro centrifuge tubes containing EDTA. Plasma samples were obtained by blood centrifugation at 3000 revolutions/min, for 15 min at 4°C. Samples were stored at –20°C until further analysis. The concentration of corticosterone in plasma was determined using commercial ELISA kits (‘HEMA’, Russia). Detection levels were 5 nmol/l according to the manufacturer.
Experimental design
C57/BL6/J as well as TRPV1 KO mice were randomly divided into two groups and deprived of food at 11:00 am. Next day (at 11:00 am) one group of C57/BL6/J as well as TRPV1 KO mice was given IM, two control groups were given its vehicle.
In experiment 1 we compared the effect of IM on the gastric mucosa in C57/BL6/J and TRPV1 KO mice. In experiment 2 we compared the effect of IM on the small intestinal mucosa in C57/BL6/J and TRPV1 KO mice.
Both vehicle- and IM-treated groups were decapitated either 4 hours after injection (experiment 1) or 24, 48 or 72 hours after the injection (experiment 2). Then, the stomach and the small intestine were removed; the length of the small intestine was measured. The stomach and the small intestine were separated, opened and photographed for examination of mucosal injury. Samples for histological analysis were also collected: 5 pieces (0.5 mm length) of the central part of the small intestine (jejunum). Blood samples were taken from trunk vessels and processed for storing.
Body weight was monitored in each of experiments. Mice were quickly weighted before fasting, after fasting (before injection) and before decapitation.
Somatic pain sensitivity was tested in IM-treated C57/BL6/J as well as TRPV1 KO mice. In experiment 1 tail flick latencies were measured under circumstances of gastric injury (4 hours after IM injection). In experiment 2 tail flick latencies were measured before IM injection (baseline tail flick latency) and 48 and 72 hours after IM injection.
Data and statistical analysis
Data was expressed as mean ± S.E.M. Data was analyzed with ANOVA module of the MedCalc Version 12.7.0.0. (Statictics for biomedical research, MedCalc Software, Belgium). Statistical significances were tested by one or two-way repeated measures ANOVA (factors: group and time), followed by a post hoc Turkey-Kramer test. When Levene’s test for homogeneity of variances was significant, nonparametric Kruskell-Wallis test was used. A student t-test was applied to analyze corticosterone levels. In each case, the required level for significance was considered to be P < 0.05.
RESULTS
Effect of indomethacin on the stomach
IM administration caused formation of hemorrhagic erosions in the glandular stomach in pre-starving animals. Gastric erosions were visible 4 hours after IM injection in both C57BL6/J and TRPV1 KO mice (Fig. 1A). However, area of gastric damages was smaller (P < 0.02) in TRPV1 KO mice than C57BL6/J mice.
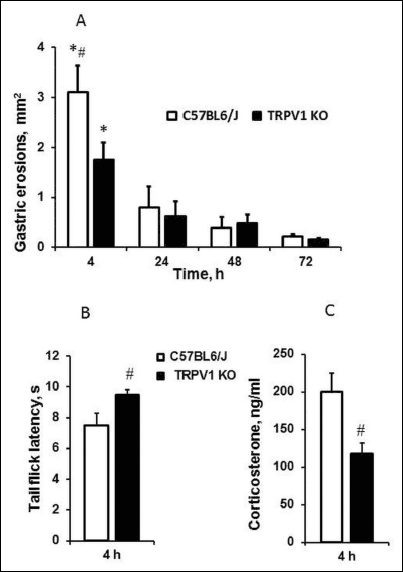 |
Fig. 1. Effect of indomethacin (IM) (35 mg/kg, subcutaneously) on gastric mucosa (A), somatic pain sensitivity (B) and plasma corticosterone levels (C). Plasma corticosterone levels were measured immediately after terminating of tail flick latency testing 4 hours after IM injection. Significant differences at P < 0.05 * versus 24, 48 ad 72 hours; # versus TRPV1 KO mice; n = 6 – 15 per group. |
Somatic pain sensitivity and corticosterone levels were also differed between C57BL6/J and TRPV1 KO mice under circumstances of formation of IM-induced gastric injury. TRPV1 KO mice were more tolerant to pain stimulation than C57BL6/J mice. They demonstrated an increase of tail flick latencies induced by tail thermal stimulation (P < 0.05) suggesting the attenuation of somatic pain sensitivity (Fig. 1B). Baseline tail flick latencies before injection were 5.68 ± 0.26 s (n = 11) in C57BL6/J mice and 7.32 ± 0.5 s (n = 13) in TRPV1 KO. We have showed that TRPV1 KO mice with less pain sensitivity had lower plasma corticosterone levels after IM administration than C57BL6/J mice (P < 0.05) (Fig. 1C). However, corticosterone levels after vehicle administration at 4 h were significantly (P < 0.05) higher in TRPV1 KO mice (110.7 ± 13.82 ng/ml, n = 19) than in C57BL6/J mice (75.33 ± 10.54 ng/ml, n = 10).
The formation of gastric erosions was followed by their healing within 72 hours that was confirmed by a decrease in the area of gastric injury (Fig. 1A) as well as histological changes (Fig. 2). Indeed, the areas of gastric damages at the 24, 48 and 72 hours after IM injection were significantly (P < 0.05) lesser than at the 4 hours in both of C57BL6/J and TRPV1 KO mice (Fig. 1A). There were no differences in area of gastric erosion between C57BL6/J and TRPV1 KO mice in the healing period of gastric injury (24, 48 or 72 hours after IM injection). There were no gastric erosions after starvation before IM or vehicle injection and also 24, 48, 72 hours after vehicle injection.
Effect of indomethacin on the small intestine
Healing of IM-induced gastric erosions was accompanied by an appearance of the small intestinal injury (Figs. 3 and 4). Small intestinal damages were detectable 24, 48 and 72 hours after IM injection in both of C57BL6/J and TRPV1 KO mice. In C57BL6/J mice the total area of injury was constant and did not alter within 72 hours after IM injection (Fig. 3A). In TRPV1 KO mice the total area of injury 24 hours after IM injection was the same as in C57BL6/J mice (Fig. 3A). Then, 48 and 72 hours after IM injection, areas of injury dramatically increased ( P < 0.05) and were significantly (P < 0.05) greater than in C57BL6/J mice. Four hours after IM administration there were no lesions in small intestine at all or they had changes that did not reach a significant level in C57BL6/J (19.36 ± 4.7 mm2, n = 9) as well as TRPV1 KO (10.22 ± 3.96 mm2, n = 16) mice compared with vehicle treated animals.
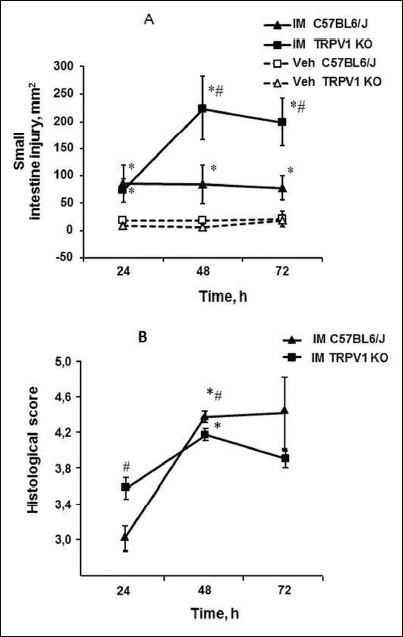 |
Fig. 3. Effect of indomethacin (35 mg/kg, subcutaneously) on small intestinal mucosa. Indomethacin-induced injury was evaluated by macroscopically (A) as well as histologically (B) 24, 48 or 72 hours after indomethacin injection. Significant differences at P < 0.05 (A): * versus corresponding vehicle group; # between TRPV1 KO and C57BL6/J mice; n = 6 – 14 per group; (B): * versus 24 h; # between TRPV1 KO and C57BL6/J mice; n = 6 – 14 per group. |
Histological analysis revealed the differences between C57BL6/J and TRPV1 KO mice 24 hours after IM injection (Fig. 3B). Although the total areas of intestinal injury did not differ in C57BL6/J and TRPV1 KO mice, histological score was significantly (P < 0.05) higher in TRPV1 KO than C57BL6/J mice. Then, at 48 hours and 72 hours after IM injection histological score was increased not only in TRPV1 KO, but also in C57BL6/J mice (P < 0.05), although the area of injury did not change in C57BL6/J mice.
The pathological action of IM was apparent not only as small intestinal damage, but also as a decrease in the small intestine length (Fig. 5). Both of C57BL6/J and TRPV1 KO mice have demonstrated the shortening of the small intestine at 48 h (P < 0.05) and 72 h (P < 0.05) after IM injection as compared to vehicle-treated animals (Fig. 5). However, the shortening of the small intestine 72 h after IM injection was more expressed in TRPV1 KO mice than in C57BL6/J mice: the length of the small intestine was significantly shorter (P < 0.05) in TRPV1 KO mice compared to C57BL6/J mice (Fig. 5).
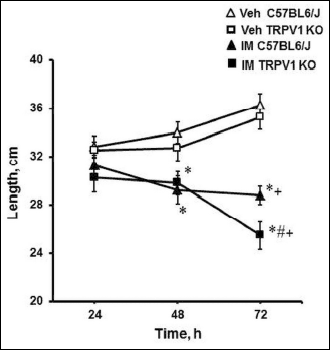 |
Fig. 5. Effect of indomethacin (35 mg/kg, subxcutaneously) on the small intestine length in C57BL6/J and TRPV1 KO mice. Significant differences at P < 0.05 * versus corresponding vehicle group; # between indomethacin-treated TRPV1 KO and C57BL6/J groups; + versus 24 h; n = 4 – 15 per group. |
Effect of indomethacin on body weight
Starvation during 24 h before IM or its vehicle administration caused a decrease in body weights in both of C57BL6/J and TRPV1 KO mice (P < 0.05) (Fig. 6). Then, after refeeding, vehicle-treated mice had recovered their body weight, whereas IM-treated mice continued to lose it gradually. Most body weight loss caused by IM was observed 72 hours after IM injection in the both of C57BL6/J (Fig. 6A) and TRPV1 KO mice (Fig. 6B). At this time point body weights were significantly (P < 0.05) lower compared to their initial weight (before starvation) as well as weight after starvation and 48 hours after refeeding. Body weights of vehicle-treated animals 48 and 72 h after injection were significantly (P < 0.05) greater compared to corresponding weights in IM-treated C57BL6/J (Fig. 6A) as well as TRPV1 KO mice (Fig. 6B). It should be noted that TRPV1 KO mice gained their weight slower than C57BL6/J mice: their body weights 48 hours after refeeding was significantly less than corresponding weights in C57BL6/J mice (P < 0.05).
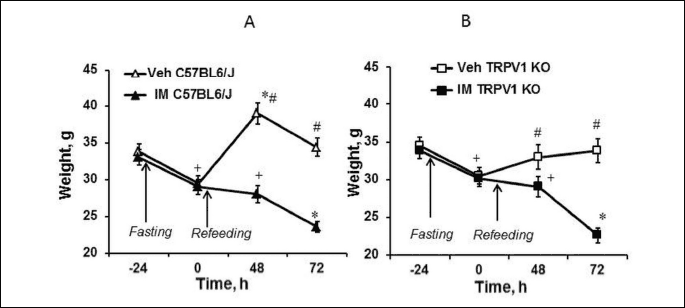
Effect of indomethacin on plasma corticosterone levels
In C57BL6/J mice IM-induced small intestinal injury was accompanied by an increase in plasma corticosterone levels at 24 h (P < 0.01) and 48 h (P < 0.01) compared to vehicle-treated animals (Fig. 7A). However, we did not discover any differences between plasma corticosterone levels in vehicle- and IM-treated TRPV1 KO mice (Fig. 7A). The differences between C57BL6/J and TRPV1 KO mice were more visible when we have compared relative changes in corticosterone levels (Fig. 7B). C57BL6/J mice had plasma corticosterone level elevation at the 24 (P < 0.02) and 48 h (P < 0.05) after IM injection, however there were no significant changes in plasma corticosterone levels in TRPV1 KO mice. IM-induced increase of corticosterone levels disappeared 72 hours after IM injection (P < 0.02) and did not differ from corresponding levels in TRPV1 KO mice as well as vehicle-induced levels. There are no significant differences between vehicle-induced levels in C57/BL6/J and TRPV1 KO mice at 24, 48 and 72 h (Fig. 7A).
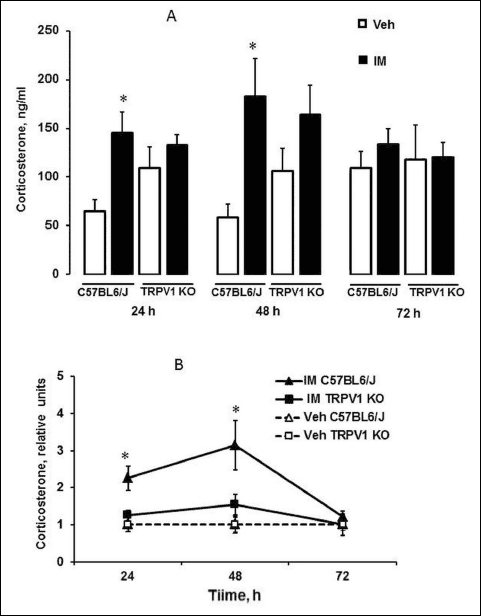 |
Fig. 7. Plasma corticosterone levels after indomethacin (vehicle) administration in C57BL6/J and TRPV1 KO mice. Plasma corticosterone levels were given in absolute (A) as well as relative units (B). The relative plasma corticosterone levels were calculated as ratio of the absolute indomethacin-induced corticosterone levels for each mouse to the corresponding mean vehicle-induced corticosterone levels, which were taken as 1 in both C57BL6 and TRPV1 KO mice. Significant difference at P < 0.05: * (A) versus corresponding vehicle group; * (B) versus corresponding IM-treated TRPV1 KO group and vehicle-treated groups; n = 3 – 15 per group. |
Effect of indomethacin on somatic pain sensitivity
We have shown that TRPV1 KO mice were more tolerant to tail thermal stimulation than C57BL6/J mice (Fig. 8). Both baseline tail flick latencies (before IM injection) as well as tail flick latencies 48 h and 72 hours after IM injection were higher in TRPV1 KO than in C57BL6/J mice (P < 0.02).
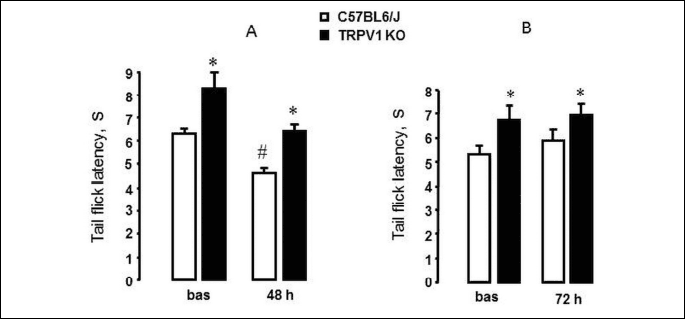
The formation of IM-induced small intestinal damages in C57BL6/J mice was accompanied by a decrease in tail flick latencies (P < 0.05) 48 hours after IM injection compared to their baseline latencies (Fig. 8A), whereas tail flick latencies 72 hours after IM injection were not different from their baseline latencies (Fig. 8B). At the same time in TRPV1 KO mice we did not observe significant changes neither 48 h (Fig. 8A) nor 72 h (Fig. 8B) after IM injection compared to their baseline latencies.
Effect of indomethacin-induced injury on animal survival
Pathological changes in the small intestine caused by IM resulted in the death of some mice in C57BL6/J as well as TRPV1 KO mice. The mortality was maximal 72 hours after IM in both of C57BL6/J and TRPV1 KO mice. However, mortality was higher in TRPV1 KO mice. Indeed, 1 of 16 mice were died for 48 h after IM injection and 5 of 11 mice – for 72 h in TRPV1 KO group whereas 2 of 16 mice were died for 48 h and 2 of 11 mice – for 72 h in C57BL6/J group. Nobody died in IM-treated groups 4 and 24 hours after its injection.
DISCUSSION
According to the data obtained in TRPV1 KO mice gastric mucosa is less vulnerable to ulcerogenic action of IM compared to C57BL6/J mice; however, their small intestinal mucosa was more vulnerable to IM action. The findings suggest that deletion of TRPV1 receptors leads to protection of gastric mucosa against ulcerogenic action of IM and to aggravation of IM-induced injury in the small intestine.
The involvement of TRPV1 receptors in both protective and pathological processes is in a good agreement with their dual sensory-efferent function (21). TRPV1 receptor stimulation resulted in the release of neuropeptides that may exert pro-inflammatory as well as anti-inflammatory or analgesic action (9-11).
TRPV1 KO mice demonstrated less IM-induced gastric injury compared to C57BL6/J mice. Similar data about less sensitivity of gastric mucosa in TRPV1 KO mice was obtained in another ulcerogenic model: acid/ethanol induced gastric injury was also significantly lesser in TRPV1 KO mice than in wild type mice (41). It seems that these data are controversial to the results demonstrated that the blockade of TRPV1 receptors by antagonist (8) or desensitization of capsaicin sensitive afferent neurons aggravates the gastric injury including IM-caused injury (31, 42). Thus, there are opposite effects of TRPV1 receptor deletion and blockade of TRPV1 receptors or desensitization of capsaicin sensitive afferent neurons on the gastric injury. It may be speculated that the deletion of TRPV1 receptors resulted in some compensatory changes developed in time and these changes contribute to the gastroprotection. This assumption is supported by fact that TRPV1 deletion causes a compensatory upregulation of several protective mechanisms in the gastric mucosa including CGRP release and cyclooxygenase 2 (COX-2) (41). Involvement of cyclooxygenase 1 (COX-1) as well as COX-2 that are responsible for produced PGs in the gastroprotection has been shown in rats with inactivation of sensory nerves by capsaicin (19). Administration of the special inhibitors of COX-1 (SC-560) and COX-2 (rofecoxib) in rats with desensitization of capsaicin sensitive afferents led to a further aggravation of gastric mucosal damage induced by stress, especially after rofecoxib administration, suggesting that COX-2 and neuropeptides released from sensory nerves may interact for the protection of the gastric mucosa (19). Additionally, TRPV1 KO mice demonstrates a reduction in myeloperoxidase activity and SP release under pathology in upper gastrointestinal tract compared with wild-type animals suggesting a decrease in the intensity of inflammatory process or may be its slower development in TRPV1 KO mice (43). However, too high levels of PGs, produced by COX-2 activation, may contribute to adverse effects, such as inflammation, increase of vessels permeability, pain and fever. Inhibition of COX-2 by IM in TRPV1 KO mice may promote other protective mechanisms in the maintenance of gastric mucosa.
Here, we confirmed our previous data that a single IM administration at the gastric ulcerogenic dose provokes pathological processes spreading from the stomach to the small intestine (38, 39). In the present study a single IM administration (35 mg/kg) caused the formation of hemorrhagic gastric erosions 4 hours after its injection, healing of which was accompanied by the small intestine injury 24, 48 ad 72 hours after IM injection in both of C57BL6/J and TRPV1 KO mice. The most marked pathological changes (an increase of histological score, shortening of the small intestine, loss of body weight) were found in the late stages of IM-induced pathological process (at 48 and 72 h after IM injection).
New result obtained here is that the deletion of TRPV1 receptors aggravates IM-induced injury in the small intestine. Although the signs of pathological process were revealed in both of C57BL6/J and TRPV1 KO. TRPV1 KO mice had greater area of injury as well as shortening of the small intestine compared to C57BL6/J mice. Additionally, TRPV1 KO mice showed lesser survival rate compared to C57BL6/J mice. The dramatic increase of total area of injury may be considered as a major factor determining survival rate.
In our study TRPV1 KO mice were more tolerant to pain stimulation and displayed higher tail flick latencies induced by thermal stimulation (reflecting decrease in somatic pain sensitivity) than C57BL6/J mice. Here, we have confirmed data of literature that TRPV1 receptors are involved in the pain regulation under thermal stimulation (5, 44). Our results are corresponding to the data showed that deletion of the TRPV1 gene or pretreatment with TRPV1 antagonists markedly attenuated thermal hyperalgesia (44-46). Taking into consideration that pain as a strong stressor activates the hypothalamic-pituitary-adrenocortical (HPA) axis, it is not surprising, that TRPV1 KO mice, lesser sensitive to pain, had lower plasma corticosterone levels than C57BL6/J mice. In TRPV1 KO mice we found less sensitivity to pain and lower corticosterone plasma levels in all point after IM injection. There was negative correlation between relative plasma corticosterone levels and IM-induced tail flick latencies (r = –0.8969,
P = 0.015). The findings are in a good agreement with the role of TRPV1 receptors in pain regulation and inflammation (5, 45).
TRPV1 receptors are also involved in visceral pain induced by pathological process in gastrointestinal tract (15, 17). Sensitization of TRPV1 receptors under inflammatory conditions underlies visceral hyperalgesia (pain), special feature of which is referring to somatic areas. It has been shown that IM-treated mice demonstrate the decrease of pain thresholds to von Frey fiber stimulation (referred hyperalgesia) compared to vehicle-treated animals both 4 and 24 hours after IM injection (47). In our study, C57BL6/J mice demonstrated a decrease of tail flick latencies only under small intestine injury (48 hours after IM injection). However, we did not find decrease in pain sensitivity under gastric injury that may be related to more intensity of IM-induced pathological process in the small intestine compared the stomach. At the same time in TRPV1 KO mice we did not find significant changes in the tail flick latencies after IM administration. It may be assumed that an increase of somatic pain sensitivity is due to visceral pain under conditions of IM-induced pathological process.
Aggravation of IM-induced injury in the small intestine in TRPV1 KO mice may suggest that TRPV1 receptors are involved in protective mechanisms that might attenuate NSAID-induced injury during prostaglandin deficiency. Based on our previous data (48-51), it may be assumed that one of protective mechanisms may be provided by compensatory action of glucocorticoids released under conditions of the small intestine injury. In the present study C57BL6/J mice that had lesser extent of the small intestine injury demonstrated higher plasma corticosterone levels 24 and 48 hours after IM injection than TRPV1 KO mice. The fact supports the involvement of glucocorticoids in the protection of the small intestinal mucosa. The results are in a good agreement with our previous data on the long-lasting HPA axis activation under circumstances of IM-induced small intestinal injury (38). Less plasma corticosterone level in TRPV1 KO mice under circumstances of small intestinal injury, when other protective mechanisms associated with prostaglandins and capsaicin sensitive neurons are impaired, may play critical role in the severity of IM-induced small intestinal injury in TRPV1 KO mice.
At the same time, 4 h after IM injection, TRPV1 KO mice showed less gastric injury compared to C57BL6/J mice, although they had lower plasma corticosterone levels. Taking into consideration a short-lasting action of ulcerogenic stimulus as well as the possible involvement of other compensatory mechanisms in TRPV1 KO mice, it may be assumed that their corticosterone levels 4 hours after IM injection were apparently sufficient for gastric mucosal protection. However, then, under long-lasting exposure of adverse factors, significance of glucocorticoids is growing and they are getting to have been necessary for further counteraction of harmful factors and, therefore, plasma corticosterone levels 48 and 72 hours after IM injection had been already insufficient for protection of small intestinal mucosa in TRPV1 KO mice.
Our data on the lacking IM-induced corticosterone rise in TRPV1 KO mice are consistent with data of lit erature that TRPV1 deletion eliminates stress-induced increase of plasma corticosterone levels and TRPV1 KO mice have lower corticosterone content than wild type animals (52). TRPV1 deletion is accompanied by reduced binding of glucocorticoids to histone deacetylase 2 promoter, that is contributing to molecular mechanisms providing stress resilient in TRPV1 KO mice (52). Lower plasma corticosterone level after IM injection and less gastric injury in TRPV1 KO mice is consistent with clinical data demonstrating that the reduction of gastric injury is accompanied by decrease in plasma cortisol levels (53). These data confirm our previous results that corticosterone rise under IM-induced prostaglandin deficiency may play compensatory gastroprotective role and may contribute to the healing process in the gastric mucosa (31, 54, 55). Glucocorticoids exert a gastroprotective effect by both maintaining local defensive factors (mucosal blood flow and mucus production) and inhibiting pathogenic elements (gastric motility and microvascular permeability) (54, 55). The gastroprotective action of glucocorticoids is also associated with maintenance of general body homeostasis, including blood glucose levels and systemic blood pressure (55).
It should be noted that capsaicin sensitive neurons may be also involved in mucosal protection through other mechanisms that are not related to TRPV1 receptors (33). A variety of TRP channels are presented in the gastrointestinal tract and may co-express by the same visceral afferents. Modulation of their functions by increased activation or sensitization as well as altered expression may also contribute to protective or pathological mechanisms in the gastrointestinal tract (15).
The results obtained suggest that TRPV1 receptors are involved in the pathological as well as protective mechanisms under circumstances of IM action on the gastrointestinal tract. Deletion of TRPV1 receptors has beneficial effect at the first stage of IM-induced pathological process for maintenance of gastric mucosa integrity but it may be harmful and aggravates pathological action of IM at the late stages of pathological process in the small intestine.
Abbreviations: CGRP, calcitonin gene-related peptide; HPA axis, hypothalamic-pituitary-adrenocortical axis; IM, indomethacin; TRPV1 KO, TRPV1 knockout; NSAIDs, nonsteroidal anti-inflammatory drugs; PGs, prostaglandins; SP, substance P; TRP channels, transient receptor potential channels; TRPV1, transient receptor potential vanilloid 1.
Acknowledgements: Study was supported by grant of Russian Science Foundation (RSF) No 14-15-00790 (study of somatic pain sensitivity under circumstances of IM-induced pathological process) and grant of RFBR No 16-04-01196a (study of indomethacin-induced injury in the stomach and the small intestine using histological methods).
Conflict of interests: None declared.
REFERENCES
- Srinivasan K. Biological activities of red pepper (Capsicum annuum) and its pungent principle capsaicin. Crit Revi Food Sci Nutr 2016; 56: 1488-1500.
- Patowary P, Pathak MP, Zaman K, Raju PS, Chattopadhyay P. Research progress of capsaicin responses to various pharmacological challenges. Biomed Pharmacother 2017; 96: 1501-1512.
- Fattori V, Hohmann M, Rossaneis A, Pinho-Ribeiro F, Verri W. Capsaicin: current understanding of its mechanisms and therapy of pain and other pre-clinical and clinical uses. Molecules 2016; 21: 844. doi: 10.3390/molecules21070844
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997; 389: 816-824.
- Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000; 288: 306-313.
- Szolcsanyi J. Hot target on nociceptors: perspectives, caveats and unique features. Br J Pharmacol 2008; 155: 1142-1144.
- Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev 1999; 51: 159-212.
- Gazzieri D, Trevisani M, Springer J, et al. Substance P released by TRPV1-expressing neurons produces reactive oxygen species that mediate ethanol-induced gastric injury. Free Radic Biol Med 2007; 43: 581-589.
- Gouin O, L’Herondelle K, Lebonvallet N, et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell 2017; 8: 644-661.
- Petho G, Bolcskei K, Furedi R, et al. Evidence for a novel, neurohumoral antinociceptive mechanism mediated by peripheral capsaicin-sensitive nociceptors in conscious rats. Neuropeptides 2017; 62: 1-10. doi: 10.1016/j.npep.2017.02.079
- Szolcsanyi J, Pinter E, Helyes Z, Petho G. Inhibition of the function of TRPV1-expressing nociceptive sensory neurons by somatostatin 4 receptor agonism: mechanism and therapeutical implications. Curr Top Med Chem 2011; 11: 2253-2263.
- Ward SM, Bayguinov J, Won KJ, Grundy D, Berthoud HR. Distribution of the vanilloid receptor (VR1) in the gastrointestinal tract. J Comp Neurol 2003; 465: 121-135.
- Yu X, Yu M, Liu Y, Yu S. TRP channel functions in the gastrointestinal tract. Semin Immunopathol 2016; 38: 385-396.
- Christianson JA, McIlwrath SL, Koerber HR, Davis BM. Transient receptor potential vanilloid 1-immunopositive neurons in the mouse are more prevalent within colon afferents compared to skin and muscle afferents. Neuroscience 2006; 140: 247-257.
- Balemans D, Boeckxstaens GE, Talavera K, Wouters MM. Transient receptor potential ion channel function in sensory transduction and cellular signaling cascades underlying visceral hypersensitivity. Amer J Physiol Gastrointest Liver Physiol 2017; 312: G635-G648.
- Beckers AB, Weerts ZZ, Helyes Z, Masclee AA, Keszthelyi D. Review article: transient receptor potential channels as possible therapeutic targets in irritable bowel syndrome. Aliment Pharmacol Ther 2017; 46: 938-952.
- Lapointe TK, Basso L, Iftinca MC, et al. TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Amer J Physiol Gastrointest Liver Physiol 2015; 309: G87-G99.
- Holzer P. Efferent-like roles of afferent neurons in the gut: Blood flow regulation and tissue protection. Auton Neurosci 2006; 125: 70-75.
- Kwiecien S, Konturek PC, Sliwowski Z, et al. Interaction between selective cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress J Physiol Pharmacol 2012; 63: 143-151.
- Mozsik G, Szolcsanyi J, Racz I. Gastroprotection induced by capsaicin in healthy human subjects. World J Gastroenterol 2005; 11: 5180-5184.
- Szolcsanyi J, Bartho L. Capsaicin-sensitive afferents and their role in gastroprotection: An update. J Physiol (Paris) 2001; 95: 181-188.
- McVey DC, Vigna SR. The capsaicin VR1 receptor mediates substance P release in toxin A-induced enteritis in rats. Peptides 2001; 22: 1439-1446.
- McVey DC, Schmid PC, Schmid HH, Vigna SR. Endocannabinoids induce ileitis in rats via the capsaicin receptor (VR1). J Pharmacol Exp Ther 2003; 304: 713-722.
- Garcia-Rayado G, Navarro M, Lanas A. NSAID induced gastrointestinal damage and designing GI-sparing NSAIDs. Exper Rev Clin Pharmacol 2018; 11: 1031-1043.
- Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology 2005; 128: 1172-1178.
- Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatolol 2005; 3: 55-59.
- Higashimura Y, Naito Y, Takagi T, et al. Preventive effect of agaro-oligosaccharides on non-steroidal anti-inflammatory drug-induced small intestinal injury in mice. J Gastroenterol Hepatol 2014; 29: 310-317.
- Satoh H, Inada I, Hirata T, Maki Y. Indomethacin produces gastric antral ulcers in the refed rat. Gastroenterology 1981; 81: 719-725.
- Kim EK, Cho JH, Jeong AR, et al. Anti-inflammatory effects of simvastatin in nonsteroidal anti-inflammatory drugs-induced small bowel injury. J Physiol Pharmacol 2017; 68: 69-77.
- Abdel Salam OM, Szolcsanyi J, Mozsik G. The indomethacin-induced gastric mucosal damage in rats. Effect of gastric acid, acid inhibition, capsaicin-type agents and prostacyclin. J Physiol (Paris) 1997; 91: 7-19.
- Filaretova L, Bobryshev P, Bagaeva T, Podvigina T, Takeuchi K. Compensatory gastroprotective role of glucocorticoid hormones during inhibition of prostaglandin and nitric oxide production and desensitization of capsaicin-sensitive sensory neurons. Inflammopharmacology 2007; 15: 146-153.
- Larauche M, Anton PM, Peiro G, Eutamene H, Bueno L, Fioramonti J. Role of capsaicin-sensitive afferent nerves in different models of gastric inflammation in rats. Auton Neurosci 2004; 110: 89-97.
- Sano T, Utsumi D, Amagase K, et al. Lafutidine, a histamine H2 receptor antagonist with mucosal protective properties, attenuates 5-fluorouracil-induced intestinal mucositis in mice through activation of extrinsic primary afferent neurons. J Physiol Pharmacol 2017; 68: 79-90.
- Szabo A, Helyes Z, Sandor K, et al. Role of transient receptor potential vanilloid 1 receptors in adjuvant-induced chronic arthritis: in vivo study using gene-deficient mice. J Pharmacol Exper Ther 2005; 314:111-119.
- Wallace JL. NSAID gastropathy and enteropathy: distinct pathogenesis likely necessitates distinct prevention strategies. Brit J Pharmacol 2012; 165: 67-74.
- Brzozowski T. Nonsteroidal anti-inflammatory drug-induced experimental gastropathy: is gastric acid the major trigger? Clin Exper Pharmacol Physiol 2010; 37: 651-653.
- Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology 2008; 135: 41-60.
- Filaretova LP, Bagaeva TR, Zelena D. The healing of NSAID-induced gastric leasions may be followed by small intestinal and cardiovascular side effects. J Physiol Pharmacol 2011; 62: 619-625.
- Yarushkina NI, Bagaeva TR, Filaretova LP. Somatic pain sensitivity in rats exposed to the harmful actions of indomethacin on the gastrointestinal tract. Neurosci Behav Physiol 2015; 45: 780-787.
- Tanigawa T, Watanabe T, Otani K, et al. Rebamipide inhibits indomethacin-induced small intestinal injury: Possible involvement of intestinal microbiota modulation by upregulation of a-defensin 5. Eur J Pharmacol 2013; 704: 64-69.
- Akiba Y, Takeuchi T, Mizumori M, Guth PH, Engel E, Kaunitz JD. TRPV-1 knockout paradoxically protects mouse gastric mucosa from acid/ethanol- induced injury by upregulating compensatory protective mechanisms. Gastroenterology 2006; 130 (Suppl. 2): A-106.
- Peng J, Li YJ. The vanilloid receptor TRPV1: Role in cardiovascular and gastrointestinal protection. Eur J Pharmacol 2010; 627: 1-7. doi: 10.1016/j.ejphar.2009.10.053
- Fujino K, de la Fuente SG, Takami Y, Takahashi T, Mantyh CR. Attenuation of acid induced oesophagitis in VR-1 deficient mice. Gut 2006; 55: 34-40.
- Davis JB, Gray J, Gunthorpe MJ, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000; 405: 183-187.
- Willis WD. The role of TRPV1 receptors in pain evoked by noxious thermal and chemical stimuli. Exp Brain Res 2009; 196: 5-11.
- Bolcskei K, Helyes Z, Szabo A, et al. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain 2005; 117: 368-376.
- Hummel M, Cummons T, Lu P, et al. Pain is a salient ‘stressor’ that is mediated by corticotropin-releasing factor-1 receptors. Neuropharmacology 2010; 59: 160-166.
- Filaretova LP, Filaretov AA, Makara GB. Corticosterone increase inhibits stress-induced gastric erosions in rats. Am J Physiol 1998; 274: G1024-G1030.
- Filaretova L, Bagaeva T. The realization of the brain-gut interactions with corticotropin-releasing factor and glucocorticoids. Curr Neuropharmacol 2016; 14: 876-881.
- Filaretova L, Podvigina T, Bagaeva T, Bobryshev P, Takeuchi K. Gastroprotective role of glucocorticoid hormones. J Pharmacol Sci 2007; 104: 195-201.
- Filaretova L. Gastroprotective role of glucocorticoids during NSAID-induced gastropathy. Curr Pharm Des 2013; 19: 29-33.
- Wang SE, Ko SY, Jo S, et al. TRPV1 regulates stress responses through HDAC2. Cell Rep 2017; 19: 401-412.
- Lechin F, van der Dijs B, Rada I, et al. Plasma neurotransmitters and cortisol in duodenal ulcer patients. Role of stress. Dig Dis Sci 1990; 35: 1313-1319.
- Filaretova L, Bagaeva T, Makara GB. Aggravation of nonsteroidal antiinflammatory drug gastropathy by glucocorticoid deficiency or blockade of glucocorticoid receptors in rats. Life sci 2002; 71: 2457-2468.
- Filaretova L, Podvigina T, Bagaeva T, Makara G. Gastroprotective action of glucocorticoids during the formation and the healing of indomethacin-induced gastric erosions in rats. J Physiol (Paris) 2001; 95: 201-208.
A c c e p t e d : December 30, 2018