ASSOCIATION BETWEEN MICROVESICLES BEARING MONOMERIC C-REACTIVE PROTEIN AND PLATELET REACTIVITY. RELATIONSHIP WITH LOW RESPONSE TO ANTIPLATELET DRUGS?
INTRODUCTION
In the pathogenesis of cardiovascular diseases, key roles are played by coagulation disorders and hypercoagulability, mainly associated with platelet activation, aggregation, and vascular endothelial dysfunction. Excessive activation of platelets, leading to lumen thrombosis, can occur with a background of atherosclerosis in the course of acute coronary syndrome or cerebral ischemic events and is one of the major factors affecting morbidity and mortality (1, 2). Research is ongoing on new molecular species which might interfere with both previously known and new biochemical pathways which lead to the activation and aggregation of platelets. Apart from having a well-documented hemostatic function, blood platelets can be active components of various other processes. Platelets by expressing certain molecules and changing the chemotactic, adhesive, and proteolytic properties of leukocytes and endothelium can initiate an inflammatory reaction in vessel walls (3, 4). Furthermore, inflammation can have a pro-thrombotic effect by inducing platelet activation, with increased platelet reactivity secondary to interleukin 6 induced hepatic thrombopoietin production. Platelets themselves have been shown to increase levels of several growth factors and inflammatory mediators (5-7). Circulating membrane microvesicles (MVs) could potentially link platelet hemostatic processes with the immune response, and it is estimated that almost a quarter of the pro-inflammatory activity of activated platelets is associated with platelet MVs generation, with strong pro-inflammatory and pro-thrombotic properties (8). Evidence is growing that suggests close links between inflammation and thrombosis, with the activation of alternative thrombotic pathways (9). Moreover, many studies have demonstrated that, in cardiovascular diseases, circulating pentameric C-reactive protein (pCRP) can dissociate into modified or monomeric CRP (mCRP) on activated platelets and the surface of circulating MVs, and this could potentially accelerate thrombogenesis and link platelet hemostatic processes with the immune response (10-12).
Properly conducted treatments, with appropriately selected doses of drugs, can help prevent undesirable activation and aggregation of platelets and inhibit inflammation progression in most patients, resulting in a limited blood clot formation. Effective anti-aggregation therapy reduces the incidence of heart attacks, strokes, and, ultimately, revascularization procedures and/or death. Current treatment guidelines for patients with ischemic heart disease, strokes, and critical limb ischemia recommend dual anti-platelet therapy (DAPT): a combination of acetylsalicylic acid (ASA) and one of the P2Y purinoceptor 12 (P2Y12) inhibitors; plus invasive treatment where appropriate, including percutaneous coronary intervention (PCI) (12). An important clinical problem may be a lack of effectiveness of antiplatelet therapy in some patients i.e., limited inhibition of platelet aggregation despite the use of antiplatelet drugs, often associated with an increased thrombotic risk (13). It has been suggested that various mechanisms may contribute to low response to antiplatelet agents, and one possible important factor is a pro-inflammatory status with profuse production of inflammatory molecules. This review aims to overview the possible relationships between the monomeric form of C-reactive protein, membrane microvesicles, platelet activation, and a low response to antiplatelet drugs.
FORMATION AND BIOLOGICAL FUNCTION
OF PLATELET MICROVESICLES
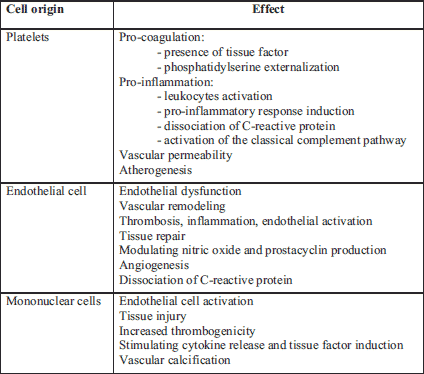
Microvesicles, exosomes, and apoptotic bodies belong to the family of extracellular vesicles (EVs) (14). The presented paper focused on platelet MVs that are characterized by their pro-coagulant activity and might reveal different roles in health and diseases. Microvesicles are small plasma membrane vesicles (0.05 – 1.00 µm in diameter) shed from various cells/platelets upon activation by factors such as serine proteases, pro-inflammatory cytokines, growth factors, and/or oxidative stress markers or apoptosis (15). Microvesicles can be generated from any cell, and the existence of microvesicles of platelet, endothelial, or monocytic origin has been demonstrated (Table 1) (15-17). However, platelet MVs make up 60 – 90% of all microvesicles in circulation (18). Microvesicles contain bioactive phospholipids, cytoplasmic components, and the surface antigens of their parent cells, owing to which their origin can be determined together with the type of stimulus causing the release. Flow cytometry is the most commonly used technique to assess MVs, allowing both enumeration and MVs cellular origin characterization. Endothelial microvesicles are recognized by adhesion molecules such as ICAM-1 (CD54), PECAM 1 (CD31), endoglin (CD105), E-selectin (CD62E), and integrins (αvβ3-CD51/61). In turn, platelet microvesicles, which have many different receptors on the membrane surface, including adhesive proteins, are recognized by CD41 (glycoprotein IIa/IIIb), CD61, CD62P (P-selectin), glycoproteins Ib, Ia/IIa, and sphingolysine, arachidonic acid (AA), and bioactive lipids (19, 20).
The exact mechanism for platelet MVs generation remains to be clearly defined. However, possible mechanisms have been identified: platelet activation induced by soluble agonists, shear stress, and glycoprotein IIb/IIIa signaling (21, 22). Besides, the generation of platelet MVs in vitro has been observed after chemical and physical platelet activation with agonists (e.g., thrombin, collagen), phorbol esters, and high shear stress or contact with artificial surfaces and complement (23). Calcium ions in the cell cytoplasm play a key role in the formation of all types of microvesicles. An increase in the intracellular concentration of calcium ions secreted by the cytoplasmic reticulum occurs during cell activation in response to many different factors, including free radicals, increased shear strength, adenosine diphosphate (ADP) secreted by activated platelets, and the expression of CD40 (CD40L) on T lymphocytes (24). This leads to the activation of concentration-dependent enzymes, such as gelsolin, which facilitate the separation of actin fibers from the platelet cytoskeleton (25); aminophospholipid translocase, which transports aminophospholipids from the plasma membrane into the cell (26); floppase, responsible for the transport of phospholipids from the inner lipid layer to the outside (27); calpain, which destroys the actin fibers of the cytoskeleton (28), and scramblase, which affects the transmembrane transport of phospholipids (29). Enzyme activation contributes to a loss of the asymmetric distribution of phospholipids in the plasma membrane, in which phosphatidylserine (PS) and phosphatidylethanolamine (PE) are mainly present in the inner cytoplasmic layer, and phosphatidylcholine (PC) and sphingomyelin (SM) in the outer lipid layer of the bilayer. The process of loss of asymmetry resulting from the transfer of phosphatidylserine and phosphatidylethanolamine to the outer layer of the cell membrane and the simultaneous destabilization of the cytoskeleton allows for the formation and secretion of microvesicles, and these phospholipids also impart a prothrombotic character to microvesicles (24).
Using proteomic and functional studies, platelet microvesicles from activated platelets were divided into heterogeneous size classes that differ significantly in terms of the content of growth factors, chemokines, and cell membrane receptors and probably have different functions in the blood (30, 31). Interestingly, on the surface of platelet MVs, there were many receptors and bioactive molecules also present in platelets. The receptors involved in primary homeostases, such as glycoprotein IIb/IIIa, P-selectin, or von Willebrand factor (vWF), may also be expressed on the surface of microvesicles (32). Procoagulation factors, chemokines, and cytokines located on the surface of platelet MVs allow microvesicles to influence pathways in inflammatory processes, atherosclerosis, and thrombosis.
It has been shown that contact of platelet microvesicles with target cells may result in monocyte chemotaxis, stimulation of cytokine secretion, activation of endothelial cells, and increased expression of tissue factor on the surface of endothelial cells (33). Platelet microvesicles stimulate the phagocytic activity of granulocytes by increasing the expression of the adhesive molecule CD11b on them (34). Microvesicles released from platelets are also involved in the upregulation of cyclooxygenase-2 (COX-2)-dependent F2-isoprostane formation in monocytes and endothelial cells, involving the transcellular metabolism of arachidonic acid. The resulting F2-isoprostane can modulate adhesive reactions and platelet activation induced by low levels of other agonists. Agonists that induce platelet activation also activate the platelet nicotinamide adenine dinucleotide phosphate oxidase (NADPH) isoform. In turn, the production of superoxide anions by NADPH oxidase on platelets enhances the recruitment of platelets into the area of a growing thrombus (35). There is now growing evidence that platelet MVs can directly regulate the immune response by carrying immunoglobulins, antigens, cytokines, and chemokines (36). The pro-inflammatory activity of platelet MVs can be demonstrated through their interaction with monocytes and neutrophils. Platelet MVs bind to leukocytes and form aggregates and can induce monocytes to release inflammatory mediators, including IL-1β, tumor necrosis alpha (TNF-α), monocyte chemotactic protein-1 (MCP-1), and matrix metalloproteinase 9 (MMP-9), which in turn, intensify the migration of monocytes (37). It is estimated that the surface of microvesicles is endowed with an average of 50 to even 100 times greater pro-inflammatory properties when computed per unit of surface area than platelets (38). Confirmation of a link between microvesicles and inflammatory processes comes from their increased levels in many prothrombotic and inflammatory disorders. An increased number of microvesicles are found in cardiovascular diseases and cancer, as well as in hemophilia and malaria. Microvesicles have also been found in low concentrations in the plasma under physiological conditions. Deficiencies in MVs generation lead to bleeding disorders with isolated prolonged bleeding time, which may be related to a deficiency in scramblase - an enzyme necessary for the formation of microvesicles (39).
The microvesicles generated during inflammation are of particular interest because they can favor the formation of pro-aggregational mCRP. Possibly, on the surface of activated platelets or circulating MVs, native pentameric CRP may dissociate to monomeric CRP, which then exerts pro-inflammatory effects by activating blood cells and promoting an inflammatory response. Monomeric CRP presence on MVs is reportedly due to the dissociation of pCRP in the microvesicles' lipid membranes (9, 11, 40). Strong correlation between serum IL-6 and CRP concentrations and platelet MVs and endothelial MVs levels were demonstrated in patients after myocardial infarction and ischemic heart disease. In contrast, no such correlations were found for microvesicles of lymphocytic and monocytic origin (41, 42). Moreover, it has been shown that MVs were increased in coronary heart disease patients (CHD) compared with non-CHD patients, with the number of platelet MVs and endothelial MVs higher in acute coronary syndrome (ACS) patients than in stable angina patients (43). Higher levels of mCRP were detected on MVs in sera of patients presenting with ST-elevation MI compared to a group who had undergone PCI (44). These results indicate that MVs play physiological roles in processes such as thrombosis and coagulation and have also been associated with vascular dysfunction and diseases such as atherosclerosis (15, 45).
C-REACTIVE PROTEIN - MONOMERIC
AND NATIVE PENTAMERIC FORMS
C-reactive protein is an acute-phase protein of the short pentraxin family, with a characteristic symmetrical shape. This protein is predominantly synthesized in the liver, typically during the transcriptional phase of the response to pro-inflammatory cytokines, most notably to IL-6 and to a lesser degree to IL-1 and TNF-α (46, 47). The native circulating form of CRP is pentameric, and each of the five identical 206-amino-acid subunits is non-covalently bonded around a central pore. There are many factors that can alter baseline CRP levels, including age, gender or smoking status (47, 48). To a large extent, the concentrations of CRP are also regulated by genetic factors (49). It has been postulated that CRP has a pro-inflammatory role in disease development (50). Numerous in vitro studies have demonstrated CRP’s participation in the activation of endothelial cells, up-regulation of cell adhesion molecules, and the increased production of inflammatory cytokines, which are common features found in cardiovascular disease pathophysiology. Likewise, correlations between complement components and CRP confirmed the interactions between inflammatory pathways and vascular disease (51). It was shown that increased blood levels of CRP correlated significantly with the incidence of cardiovascular complications in patients without any symptoms and in patients with unstable angina, MI, or peripheral artery disease. Moreover, elevated blood CRP concentrations are considered a risk factor for stroke and heart attack, and sudden death in patients after PCI (47).
Under physiological conditions, there are at least two forms of C-reactive protein in the organism which possess distinct antigenic, electrophoretic, and biological features: the ‘native’ pentameric isoform (pCRP) and the monomeric isoform (mCRP). A wide variety of in vitro and ex vivo monomeric CRP isoforms have been reported, ranging from in-vitro-denatured mCRP dissociated from native C-reactive protein by exposure to urea, heat, or acidic conditions, through reduced membrane-bound mCRP to a hybrid mCRP with near-native structure (11, 52, 53). As reported in the literature, platelets and platelet-derived microvesicles, and apoptotic/necrotic cells, play a key role in generating the monomeric forms of C-reactive protein (9, 42). Conformational changes are mediated by bioactive lipids on activated or damaged cells or platelets (10). Just as CRP dissociates upon binding to the membrane of activated platelets, the same process occurs after binding of C-reactive protein to the membranes of microvesicles derived from activated cells. Microvesicles, like apoptotic and activated cells, contain phospholipids on their surface, including lysophosphatidylcholine (LPC), which is not usually exposed in the outer layer of plasma membranes. The presence of mCRP on MVs has been reported to result from the dissociation of pCRP in a membrane lipid environment where altered lysophospholipids overcome electrostatic forces holding the monomer units together, leading to their dissociation. The conversion of pCRP to membrane-bound mCRP depends on LPC content, and MVs capable of converting pCRP to mCRP have elevated levels of lysophosphatidylcholine (54). The protein remains bound to the membrane and can therefore be detected in the circulating blood and transported. Most of the microvesicles involved in this process come from activated platelets. It has been shown that microvesicles with mCRP bound on their surface are able to bind activated endothelial cells and transmit pro-inflammatory signals to the endothelium itself (15).
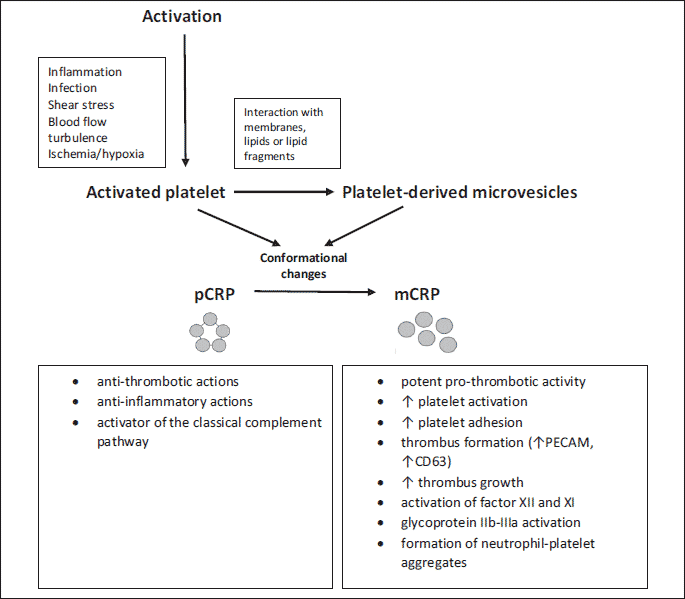
It is now apparent that the biological action of CRP is strongly dependent on its conformation, and the monomeric form has been shown to exert various effects that are distinct from those of pCRP. It is believed that pCRP is anti-inflammatory and anti-aggregating, while mCRP is pro-inflammatory and pro-aggregative (Fig. 1). Increasing experimental evidence within the literature suggests that pentameric CRP is mostly released into the circulation after an inflammatory stimulus. In contrast, mCRP is involved in the innate immune system and activates the complement cascade in angiogenesis and in thrombosis (50). It was reported that mCRP may contribute to modulating angiogenesis in acute stroke, in the atherosclerotic plaque, and in infarcted tissues (55-57). Free mCRP has not been detected in the serum, likely because of its relative aqueous insolubility. Monomeric CRP usually remains physiologically bound in tissues, including blood vessels’ walls (58, 59). However, it does appear in circulating blood but the method of its transport and influx into areas of inflammation have not been extensively studied. It is known that mCRP can be transported in the bloodstream on the surface of microvesicles that are able to convert pCRP to mCRP (60). This pool of circulating mCRP cannot be measured by routine assays to measure CRP concentration and is therefore invisible in clinical analyses. Thus, monomeric CRP, a plasma-insoluble hydrophobic isoform, by binding to microvesicles could be transported into cells where it integrates into lipid raft domains of the plasma membrane via putative cholesterol-binding domains. Thus, it has been shown that mCRP can possibly induce pro-inflammatory effects in cells such as monocytes or endothelial cells, promoting the release of pro-inflammatory cytokines, the production of reactive oxygen species and the expression of adhesion molecules, and also enhancing the production of MVs (61, 62).
It is difficult to determine the percentage of native pentameric CRP which has dissociated to the monomeric form. The routine methods of CRP measurements (turbidimetric or nephelometric) do not differentiate the two CRP isoforms, and the high-sensitivity CRP (hsCRP) test typically refers to the measurement of the pentameric form of CRP in blood serum. However, recently a highly specific ELISA assay has been developed to measure plasma and tissue levels of mCRP. Zhang et al. have quantified mCRP concentrations in patients with skin-related autoimmune disorders using a sandwich ELISA and obtained a statistically significant difference in mCRP levels compared to the control group, while the native pentameric CRP did not show such a difference (63). Wang et al., on the other hand, used a monoclonal antibody based on the C-terminal octapeptide of CRP to assess mCRP concentrations in patients with acute myocardial infarction (AMI) and showed that the antibody used recognizes mCRP from denatured pCRP but not pCRP itself (64). It is also possible to quantify the monomeric form of CRP located on platelet microvesicles by flow cytometry (19, 42, 43).
ASSOCIATION BETWEEN MONOMERIC CRP-BEARING
MICROVESICLES AND PLATELET HYPERFUNCTION
Pathophysiological associations between CRP, microvesicles, and platelet hyperfunction have been suggested, although data in this field still conflicts. The majority of MVs circulating in the blood have been found to be shed by activated platelets, which are mainly associated with platelet activation and aggregation. More and more evidence now supports the concept that microvesicles are involved in platelet function and are important regulators of hemostasis, inflammation, and angiogenesis (38, 43). It has been observed that platelet MVs can exert their effects in many ways, including triggering cascades of intercellular signaling and participating in cell-to-cell communication by transferring cytoplasmic components and surface receptors to other cell types. Due to exposure to negatively charged phosphatidylserine, platelet MVs have binding sites for high-affinity coagulation factors, including factors Va, VIIIa, and IXa (65). In addition, they show a higher surface density of factor Xa compared to activated platelets, which allows the formation of internal tenase (factor IXa/VIIIa) and prothrombinase (factor Xa/Va) complexes, thus enhancing clotting pathways both independent and dependent on tissue factor (TF) (66).
It has been shown that platelet activation enhances the release of platelet MVs. On the other hand, it has also been observed that a sub-population of platelet MVs derived from agonist-activated platelets may also contribute to pathological events (67). Thus, platelet MVs may well be both a cause and a consequence of the pathophysiology that drives various diseases. Elevated levels of platelet MVs have been observed in various conditions associated with platelet activation such as ischemic stroke, coronary artery disease, heparin-induced thrombocytopenia (HIT), hypertension, diabetes, obesity, cancer, and metastases, and a potential correlation between the number of platelet MVs and the clinical severity of the disease is suggested (68-71). As platelet microvesicles are highly prothrombotic it has been suggested that they may contribute to the development of arterial thrombosis and may provide a potential prognostic marker for atherosclerotic vascular disease (72). Platelet microvesicles with exposed P-selectin and CD63 on their surface have been shown to reflect platelet activation in peripheral arterial disease and myocardial infarction (73). Increased levels of platelet MVs have been recorded in survivors of myocardial infarction (74). A significant independent relationship has also been observed between large platelet microvesicles with plasma thrombin-antithrombin complexes and soluble CD40 ligand (sCD40L) in patients with myocardial infarction, but not in healthy subjects (74). More recent studies have also shown that internal and external carotid-artery diameters negatively correlate with the number of microvesicles derived from platelets, endothelial cells, and leukocytes. However, it is unclear whether the microvesicles contribute to the remodeling of the artery or represent the activation state of the cells from which they are derived (75).
C-reactive protein is present in the circulation, so the effect of CRP on hemostasis observed in vivo might be related to an excessive formation of modified CRP molecules. As mentioned earlier, pentameric CRP can bind to microvesicles of various origins, including platelet MVs (76). Microvesicles linked to pCRP, although not pro-inflammatory in healthy people, may exacerbate existing tissue damage by activating the classical complement pathway and enhancing leukocyte recruitment to inflamed tissues (76). The microvesicles not only bind the pentameric form of CRP, but also transform pCRP into the highly pro-inflammatory monomer of C-reactive protein that can bind to endothelial cells and generate pro-inflammatory signals (42, 77). It appears that the presence of inflammation can lead to increased formation of mCRP from pCRP in the bloodstream, thus linking thrombosis and inflammation, key events in acute coronary syndromes.
Interestingly, this concept suggests that the mere presence of CRP in plasma is not associated with platelet aggregation but that it is probably monomeric CRP that may be one of the triggers of prothrombotic status. Monomeric CRP has been detected in atherosclerosis plaques, within infarctions, in several skin-related autoimmune disorders, and bound to microvesicles in patients’ blood circulation following MI (44, 45, 59, 62). It has been noticed that the monomeric form induces platelet aggregation and activation, whereas the native pentameric form is unable to produce any effect, even at high concentrations (10, 78). After dissociation CRP is able to promote a prothrombotic state by inducing platelet activation and platelet adhesion by upregulating P-selectin. Flow cytometric analysis of platelets activated by collagen demonstrated that only mCRP significantly increased platelet-surface P-selectin expression without modifying CD63 and PAC-1 (73, 77). Furthermore, using confocal immunohistochemistry the presence of mCRP on the surface of adhered platelets and within the thrombus was reported (10, 77). Monomeric CRP was associated with a presentation of a prothrombotic phenotype, enhancing not only platelet deposition but also thrombus growth under arterial flow conditions, suggesting a stable and direct interaction between platelets and CRP (11). It has been shown that both C-reactive protein isoforms can bind to resting and activated platelets, but only activated platelets are able to dissociate pentameric CRP into monomeric CRP on their surface. On the other hand, monomeric CRP produced on the surface of adhered platelets shows prothrombotic properties and stimulates further platelet deposition. The effect of mCRP on the increase in P-selectin expression may partially explain the relationship of mCRP with thrombus enlargement (78). As a result, we are dealing with a positive feedback mechanism in which the dissociation of pentameric C-reactive protein on adjacent platelets may facilitate further activation of platelets (78). Previous studies have shown that platelets could be activated by mCRP in a static incubation system. The monomeric CRP form could even enhance platelet activation induced by DNase-I-pretreated netting neutrophils in the static incubation system. Besides, mCRP on activated platelets could enhance platelet activation and the formation of D-dimers in perfusion experiments (79). Furthermore, monomeric CRP can activate in vitro leukocytes and stimulates the generation of reactive oxygen species, expression of adhesion molecules, and cytokine and chemokine release (80). Additionally, mCRP but not pCRP can significantly increase platelet adhesion when directly incubated with blood and when immobilized on a collagen surface (11).
These observations support the role of mCRP in platelet adhesion as well as in platelet interactions that are responsible for clot enlargement and further vessel occlusion. Moreover, it has been observed that, unlike the pentameric C-reactive protein, the monomeric C-reactive protein led to conformational changes in glycoprotein IIb/IIIa in a dose-dependent manner. In turn, blocking glycoprotein IIb/IIIa on activated platelets prevented pCRP from dissociating into mCRP and inhibited the deposition of platelets in the artery wall (81). The results of many studies have highlighted the ability of platelet MVs to interact with various proteins, including CRP, to induce a prothrombotic and pro-inflammatory phenotype. Therefore, platelet MVs and CRP isoforms can play a dual role: they can elicit a pro- or anti-inflammatory response depending on the form of the inflammatory protein, the composition of the cell membrane, and the biological load carried by the platelet MVs.
LOW RESPONSE TO ANTIPLATELET DRUGS - POSSIBLE
LINKS TO MICROVESICLES BEARING MONOMERIC CRP?
Antiplatelet drugs differ in their mode of action. Glycoprotein IIb/IIIa receptor inhibitors inhibit the last stage of primary hemostasis - platelet aggregation - and acetylsalicylic acid and clopidogrel block the first stage - platelet activation. ASA is an antiplatelet drug that irreversibly inhibits platelet cyclooxygenase-1 (COX-1), an enzyme in the arachidonic acid pathway, and blocks the production of thromboxane A2 (TXA2) and adenosine diphosphoric acid (ADP). Acetylsalicylic acid irreversibly inhibits prostaglandin production by acetylation of the serine at the arachidonic acid binding site, which effectively inhibits platelet aggregation (81, 82). Clopidogrel, unlike ASA, is a prodrug - 85% of the administered dose is inactivated by plasma esterases, and only 15% undergoes a complex transformation into its active form in the liver. The active form of clopidogrel specifically and irreversibly inhibits the purinergic platelet receptor P2Y12, preventing ADP from activating platelet receptors and resulting in inhibition of platelet activation and aggregation. The effects of clopidogrel, i.e., platelet inhibition, vary between individuals and have been shown to correlate directly with cytochrome p450 activity (83, 84). Clopidogrel belongs to the second generation of thienopyridine antiplatelet agents and has been the most utilized P2Y12 inhibitor in routine clinical practice for several years. However, incremental benefits have been shown with more potent P2Y12 inhibitors, e.g., prasugrel and ticagrelor. Prasugrel is a specific, irreversible antagonist of the platelet P2Y12 ADP-receptor and belongs to the third generation of oral thienopyridine antiplatelet agents. Ticagrelor is an adenosine triphosphate (ATP) derivate and belongs to a new class of orally active antiplatelet agents that do not require metabolic activation. The thienopyridine classes inhibit the P2Y12 ADP-receptor, but ticagrelor’s linkage is completely reversible (13). Laboratory studies have shown that prasugrel and ticagrelor are associated with potent and predictable degrees of platelet inhibition compared with clopidogrel, but, on the other hand, such a significant reduction in ischemic events may lead to more bleeding complications (84).
Effectiveness of antiplatelet therapy may be limited due to low response to antiplatelet drugs that often results in the recurrence of vascular events, even with the correct treatment (85, 86). Despite the fact that significant progress has recently been made in recognizing and combatting an abnormal response to oral antiplatelet agents, it is still unknown why some patients who receive dual antiplatelet therapy do not benefit from the expected therapeutic effects. It is well known that individual responsiveness to P2Y12 inhibitors and/or aspirin varies widely among patients and is subject to intra- and inter-individual variability. The prevalence of non-responsiveness is more significant in patients treated with clopidogrel compared with prasugrel (3 – 15%) and ticagrelor (0 – 3%), and high on-treatment platelet reactivity to therapeutic response to dual antiplatelet therapy with these latter agents plays only a marginal role (87-89). This difference is likely due to different drug pharmacokinetics whereby prasugrel and ticagrelor generate active metabolites more efficiently than clopidogrel.
Factors that cause the huge variation in antiplatelet therapy response are not fully resolved. Multiple mechanisms may be responsible for low biological response to antiplatelet agents, including clinical, cellular, and genetic factors. Important factors seem to be genetic variability (e.g., polymorphisms in the genes for P2Y12, glycoprotein IIb/IIIa platelet receptors, or COX-1 and COX-2 polymorphisms) as well as drug-drug interactions. Low cytochrome P450 activity or alternative pathways of platelet activation - especially thrombin production and accelerated platelet turnover - or activation of independent P2Y12 pathways and increased ADP exposure may also lead to a differential response to antiplatelet drugs (90, 91). Increased platelet reactivity, upregulation of multiple platelet adhesion, activation and aggregation pathways, and the release of inflammatory markers and prothrombotic factors appear to be of particular importance. High baseline platelet reactivity before treatment may also contribute to decreased antiplatelet effects induced by clopidogrel or aspirin, especially in patients with acute coronary syndromes, diabetes, heart failure, and stroke (92, 93). Therefore, consideration should be given to comorbidities that may trigger a higher platelet reactivity and a variable antiplatelet response (e.g., hypertension, hyperlipidemia, heart failure, diabetes, obesity, or inflammation) (94). Our earlier work showed that diabetes with CHD weakens the response to antiplatelet drugs, especially to clopidogrel, and that hyperglycemia, hypertension, and obesity might also play important roles. Moreover, a diabetic's low response to ASA is associated with increased platelet reactivity, perhaps related to the more frequent ITGB3 PIA1 allele and increased TXB2 generation (95).
Aspirin and/or clopidogrel provide therapeutic benefits mainly due to their antiplatelet activity, but their anti-aggregation effect is not the only result of their full effectiveness. Due to the close relationship between platelet activation, thrombus formation, and inflammation, it is believed that effective platelet inhibition is associated with improved endothelial function and decreased expression of inflammatory markers. The results of clinical trials confirm that treatment with clopidogrel can reduce vascular inflammation in patients with ischemic vascular disease, especially in patients with acute coronary syndrome undergoing PCI. Clopidogrel also inhibits the release of soluble P-selectin and CD40 ligands by platelets and significantly lowers P-selectin expression (96). Moreover, there is evidence to suggest that antiplatelet efficacy may be impaired in the presence of inflammation. The association between severe atherothrombotic events and inflammation can be explained by activation of alternative thrombotic pathways that affect clotting factors, platelets, endothelial cells, and leukocytes, resulting in a decreased response to aspirin and/or clopidogrel in inflammation. The interaction of platelets, endothelial cells, and leukocytes may play a key role in the development of decreased efficacy of antiplatelet therapy by maintaining pro-inflammatory and pro-thrombotic states.
One question is whether low response to antiplatelet drugs is a cause or effect, especially in the presence of inflammation. Despite the strong evidence linking inflammation and thrombosis, the exact mechanisms by which severe inflammation may influence the response to antiplatelet agents have not yet been fully elucidated. One of the most popular inflammatory biomarkers associated with cardiovascular risk factors is CRP. Hepatic CRP production is directly related to IL-6 stimulation, and interestingly, this stimulation also follows a similar pathway to platelet production and platelet turnover during inflammation (97). When bound to platelets or platelet MVs, dissociated CRP may reduce physiological platelet agonists’ effects, inhibit platelet secretion, and enhance the antiplatelet effects of acetylsalicylic acid, indicating that the effectiveness of aspirin therapy may strongly depend upon the levels of CRP in circulation (98, 99). Moreover, it has been shown that mCRP, by activating the IIIa subunit of the platelet glycoprotein receptor, enhances platelet aggregation under the influence of ADP, which may, therefore, be a factor in a weakened response to clopidogrel (64).
Some researchers have indicated that platelet MVs are potential markers for the antiplatelet effects of both aspirin and P2Y12 inhibitors in patients with acute coronary syndromes and after PCI procedures, and previous studies have shown a reduction in platelet MVs generation in patients with acute coronary syndrome treated with aspirin and P2Y12-receptor antagonists (100-102). Clopidogrel has been shown to inhibit the release of membrane microvesicles from endothelial and platelet progenitor cells in patients with the stable coronary-artery disease (103). On the other hand, patients with high on-treatment platelet reactivity during clopidogrel treatment have elevated circulating platelet MVs levels, indicating ongoing platelet activation despite clopidogrel treatment (104). It has also been observed that a maintenance dose of 75 mg clopidogrel differentially affects platelet aggregation and platelet-derived pro-thrombotic and pro-inflammatory mediators in ACS patients within the first month of treatment, a phenomenon that is highly influenced by drug response variability. Additionally, under experimental conditions, an active metabolite of prasugrel strongly inhibited collagen-induced platelet MVs formation showing that activation of the arachidonic acid pathway and the release of ADP from delta granules amplify the release of microvesicles from platelets (105). In a very recent in vitro study, it was asserted that ticagrelor has the capability to induce inhibition of ADP-induced platelet aggregation and ADP-mediated generation of platelet MVs from activated platelets (defined as CD61(GPIIIa)/CD62P/PS triple-positive MV) (106). It has also been shown that the higher antiplatelet potency of ticagrelor and prasugrel is associated with decreased platelet MVs plasma counts (106). However, circulating platelet MVs in subjects treated with prasugrel or ticagrelor or in vivo comparisons of blood platelet MVs counts between patients on various P2Y12 antagonists have been estimated in only a few studies.
In this area of research, we have our own observations regarding the relationship between inflammation and low response to aspirin in patients with ischemic stroke. The results of our published studies have indicated a worse response to ASA treatment in patients with higher CRP levels. There was also a strong positive correlation between CRP concentrations and platelet aggregation in a group of refractory patients as measured by impedance aggregometry (107). Moreover, we noticed that with low response to ASA and/or clopidogrel in patients with stable coronary disease awaiting PCI, there was an inferior response to combined antiplatelet therapy in women than in men, potentially connected with a more pronounced pro-inflammatory status in women as determined by higher concentrations of C-reactive protein and higher numbers of leukocytes and platelets (108).
CONCLUSION
The mechanisms of CRP interaction with platelets or platelet MVs, despite many studies, are still poorly understood, partly due to at least two CRP isomeric forms. Results suggest that mCRP levels are not only more sensitive to the local status of inflammation but may also be specific to the underlying pathogenesis. Part of the variable antithrombotic action that antiplatelet agents exert in different clinical settings may be explained by a modulation in the generation of mCRP bound to platelet MVs. Therefore, further research into the role of MVs and studies on conformational changes of the CRP protein in patients with coronary artery disease may help explain the insufficient effectiveness of antiplatelet therapy. Furthermore, markers of inflammation, such as mCRP or platelet MVs, may be useful markers of antiplatelet therapy. It is suggested that circulating or microparticle-bound mCRP may provide a better diagnostic index than pCRP in myocardial infarction and peripheral artery disease. In turn, platelet microvesicles may provide a marker of disease activity or response to treatment and may be used in the prognosis of the course of various diseases.
Source of funding: None.
Conflict of interests: None declared.
REFERENCES
- van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol 2019; 16: 166-179.
- Khodadi E. Platelet function in cardiovascular disease: activation of molecules and activation by molecules. Cardiovasc Toxicol 2020; 20: 1-10.
- Steinhubl SR. Platelets as mediators of inflammation. Hematol Oncol Clin N Am 2007; 21: 115-121.
- Muhlestein JB. Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb Haemost 2010; 103: 71-82.
- Holinstat M. Normal platelet function. Cancer Metastasis Rev 2017; 36: 195-198.
- Ware J, Corken A, Khetpal R. Platelet function beyond hemostasis and thrombosis. Curr Opin Hematol 2013; 20: 451-456.
- Thachil J. Platelets in inflammatory disorders: a pathophysiological and clinical perspective. Semin Thromb Hemost 2015; 41: 572-581.
- Badimon LR, Suades E, Fuentes I, Palomo TP. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: a link between inflammation, atherosclerosis, and thrombosis. Front Pharm 2016; 7: 293. doi: 10.3389/fphar.2016.00293
- Filep JG. Platelet affects the structure and function of C-reactive protein. Circ Res 2009; 105: 109-111.
- Boncler M, Rywaniak J, Szymanski J, Potempa LA, Rychlik B, Watala C. Modified of C-reactive protein interacts with platelet glycoprotein Ibα. Pharmacol Rep 2011; 63: 464-475.
- Boncler M, Wu Y, Watala C. The multiple faces of C-reactive protein-physiological and pathophysiological implications in cardiovascular disease. Molecules 2019; 24: 2062. doi: 10.3390/molecules24112062
- Gulizia MM, Colivicchi F, Abrignani MG, et al. ESC Scientific Document Group; Faculty for approval of the Consensus Document. Consensus Document ANMCO/ANCE/ARCA/GICR-IACPR/GISE/SICOA: Long-term antiplatelet therapy in patients with coronary artery disease. Eur Heart J Suppl 2018; 20 (Suppl. F): F1-F74. doi: 10.1093/eurheartj/suy019
- Warlo EMK, Arnesen H, Seljeflot I. A brief review on resistance to P2Y12 receptor antagonism in coronary artery disease. Thromb J 2019; 17: 11. doi: 10.1186/s12959-019-0197-5
- Ridger VC, Chantal M, Boulanger CM, et al. Microvesicles in vascular homeostasis and diseases. Thromb Haemost 2017; 117: 1296-1316.
- Wu ZH, Ji CL, Li H, Qiu GX, Gao CJ, Weng XS. Membarne microparticles and diseases. Eur Rev Med Pharmacol Sci 2013; 17: 2420-2427.
- Zaldivia MTK, McFadyen JD, Lim B, Wang X, Peter K. Platelet-derived microvesicles in cardiovascular diseases. Front Cardiovasc Med 2017; 4: 74. doi: 10.3389/ fcvm.2017.00074
- Berezin EA, Berezin AA. Platelet-derived vesicles: diagnostic and predictive value in cardiovascular diseases. J Unexplored Med Data 2019; 4: 4. doi.org/10.20517/2572-8180.2019.05
- Siljander PR. Platelet-derived microparticles - an updated perspective. Thromb Res 2011; 127: 30-33.
- Baj-Krzyworzeka M, Majka M, Pratico D, et al. Platelet derivied microparticles stimulate proliferation, survival, adhesion, and chemotaxis of haemopoetic cells. Exp Hematol 2002; 30: 450-459.
- Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev 2007; 21: 157-171.
- Connor DE, Exner T, Ma DD, Joseph JE. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost 2010; 103: 1044-1052.
- Diehl P, Aleker M, Helbing T, et al. Increased platelet, leukocyte and endothelial microparticles predict enhanced coagulation and vascular inflammation in pulmonary hypertension. J Thromb Thrombolysis 2011; 31: 173-179.
- Wang Y, Zhang S, Luo L, et al. Platelet-derived microparticles regulates thrombin generation via phophatidylserine in abdominal sepsis. J Cell Physiol 2018; 233: 1051-1060.
- Morel O, Morel N, Freyssinet JM, Toti F. Platelet microparticles and vascular cells interactions: a checkpoint between the haemostatic and thrombotic responses. Platelets 2008; 19: 9-23.
- McLaughlin PJ, Gooch JT, Mannherz HG, Weeds AG. Structure of gelsolin segment 1-actin complex and the mechanism of filament serving. Nature 1993; 364: 685-692.
- Beleznay Z, Zachowski A, Devaux PF, Navazo MP, Ott P. ATP-dependent aminophospholipid translocation in erythrocyte vesicles: stoichiometry of transport. Biochemistry 1993; 32: 3146-3152.
- Connor J, Pak CH, Zwaal RF, Schroit AJ. Bidirectional transbilayer movement of phospholipid analogs in human red blood cells. Evidence for an ATP-dependent and protein-mediated process. J Biol Chem 1992; 267: 19412-19417.
- Kelton JG, Warkentin TE, Hayward CP, Murphy WG, Moore JC. Calpain activity in patients with thrombotic thrombocytopenic purpura is associated with platelet microparticles. Blood 1992; 80: 2246-2251.
- Zwaal RF, Comfurius P, Bevers EM. Mechanism and function of change in membrane-phospholipid asymetry in platelets and erythrocytes. Biochim Soc Trans 1993; 21: 248-253.
- Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and functional characterisation of platelet microparticle size classes. Thromb Haemost 2009; 102: 711-718.
- Aatonen MT, Ohman T, Nyman TA, Laitinen S, Gronholm M, Siljander PR. Isolation and characterization of platelet-derived extracellular vesicles. J Extracell Vesicles 2014: 3: 1. doi:10.3402/jev.v3.24692
- Curry N, Raja A, Beavis J, Stanworth S, Harrison P. Levels of procoagulant microvesicles are elevated after traumatic injury and platelet microvesicles are negatively correlated with mortality. J Extracell Vesicles 2014; 3: 25625. doi:10.3402/jev.v3.2562535
- Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest 1998; 102: 136-144.
- Merten M, Pakala P, Thiagarajan P, Benedict CR. Platelet microparticles promote platelet interactions with subendothelial matrix in a glycoprotein IIb/IIIa-dependent mechanism. Circulation 1999; 99: 2577-2582.
- Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357: 2482-2494.
- Cloutier N, Tan S, Boudreau LH, et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. EMBO Mol Med 2013; 5: 235-249.
- Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 2000; 95: 1317-1323.
- Shantsila E, Kamphuisen PW, Lip GY. Circulating microparticles in cardiovascular disease: implications for atherogenesis and atherothrombosis. J Thromb Haemost 2010; 8: 2358-2368.
- Rank A, Nieuwland R, Nikolajek K, et al. Hormone replacement therapy leads to increased plasma levels of platelet derived microparticles in postmenopausal women. Arch Gynecol Obstet 2012; 285: 1035-1041.
- Strasser EF, Happ S, Weiss DR, Pfeiffer A, Zimmermann R, Eckstein R. Microparticles detection in platelet products by three different methods. Transfusion 2013; 53: 156-166.
- van der Zee PM, Biro E, Trouw LA, et al. C-reactive protein in myocardial infarction binds to circulating microparticles but is not associated with complement activation. Clin Immunol 2010; 135: 490-495.
- Cui Y, Zheng L, Jiang M, et al. Circulatin microparticles in patients with coronary heart disease and its correlation with interleukin-6 and C-reactive protein. Mol Biol Rep 2013; 40: 6437-6442.
- Biasucci LM, Porto I, Di Vito L, et al. Differences in microparticle release in patients with acute coronary syndrome and stable angina. Circ J 2012; 76: 2174-2182.
- Habersberger J, Strang F, Scheichl A, et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res 2012; 96: 64-72.
- Crawford JR, Trial J, Nambi V, Hoogeveen RC, Taffet GE, Entman ML. Plasma levels of endothelial microparticles bearing monomeric C-reactive protein are increased in peripheral artery disease. J Cardiovasc Transl Res 2016; 9: 184-193.
- Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol 2001; 38: 189-197.
- Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol 2018; 9: 754. doi: 10.3389/fimmu.2018.00754
- Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 2008; 456: 989-992.
- Carlson CS, Aldred SF, Lee PK, et al. Polymorphism within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am J Hum Genet 2005; 77: 64-77.
- McFadyen JD, Kiefer J, Braig D, et al. Dissociation of C-reactive protein localizes and amplifies inflammation: evidence for a direct biological role of C-reactive protein and its conformational changes. Front Immunol 2018; 9: 1351. doi: 10.3389/fimmu.2018.01351
- Runjic F, Martinovic-Kaliterna D, Salamunic I, Kristic I, Ljubkovic M, Marinovic J. Association of anticardiolipin antibodies, complement and leptin with the severity of coronary artery disease expressed as syntax score. J Physiol Pharmacol 2020; 71: 383-388.
- Kresl JJ, Potempa LA, Anderson BE. Conversion of native oligomeric to a modified monomeric form of human C-reactive protein. Int J Biochem Cell Biol 1998; 30: 1415-1426.
- Ahrens I, Domeij H, Eisenhardt SU, Topcic D, Albrecht M, Leitner E. Opposin effects of monomeric and pentameric C-reactive protein on endothelial progenitor cells. Basic Res Cardiol 2011; 106: 879-895.
- Kiefer CR, Stock RE, Flanagan SS, Darling CE, Smith CS, Snyder LM. Early verification of myocardial ischemia with a novel biomarker of acute tissue damage: C-reactive protein fractional forms. Clin Chim Acta 2012; 413: 1536-1541.
- Slevin M, Matou-Nasri S, Turu M, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol 2010; 20: 151-165.
- Pena E, de la Torre R, Arderiu G, Slevin M, Badimon L. mCRP triggers angiogenesis by inducing F3 transcription and TF signalling in microvascular endothelial cells. Thromb Haemost 2017; 117: 357-370.
- Badimon L, Pena E, Arderiu G, et al. C-reactive protein in atherothrombosis and angiogenesis. Front Immunol 2018; 9: 430. doi: 10.3389/fimmu.2018.00430
- Diehl EE, Haines GK3rd, Radosevich JA, Potempa LA. Immunohistochemical localization of modified C-reactive protein antigen in normal vascular tissue. Am J Med Sci 2000; 319: 79-83.
- Eisenhardt SU, Habersberger J, Murphy A, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res 2009; 105: 128-137.
- Melnikov I, Kozlov S, Saburova O, et al. CRP is transported by monocytes and monocyte-derived exosomes in the blood of patients with coronary artery disease. Biomedicines 2020; 8: 435. doi:10.3390/biomedicines8100435
- Ji SR, Ma L, Bai CJ, et al. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB 2009; 23: 1806-1816.
- Li HY, Wang J, Wu YX, et al. Topological localization of monomeric C-reactive protein determines proinflammatory endothelial cell responses. JBC 2014; 289: 14283-14290.
- Zhang L, Li HY, Li W, et al. An ELISA assay for quantifying monomeric c-reactive protein in plasma. Front Immunol 2018; 9: 511. doi: 10.3389/fimmu.2018.00511
- Wang J, Tang B, Liu X, et al. Increased monomeric CRP levels in acute myocardial infarction: a possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis 2015; 239: 343-349.
- Chyrchel B, Drozdz A, Dlugosz D, Stepien EL, Surdacki A. Platelet reactivity and circulating platelet-derived microvesicles are differently affected by P2Y12 receptor antagonists. Int J Med Sci 2019; 16: 264-275.
- Sinauridze EI, Kireev DA, Popenko NY, et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost 2007; 97: 425-434.
- Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010; 327: 580-583.
- Leroyer AS, Tedgui A, Boulanger CM. Role of microparticles in atherothrombosis. J Int Med 2008; 263: 528-537.
- Suades R, Padro T, Vilahur G, Badimon L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb Haemost 2012; 108: 1208-1219.
- Campello E, Zabeo E, Radu CM, et al. Hypercoagulability in overweight and obese subjects who are asymptomatic for thrombotic events. Thromb Haemost 2015; 113: 85-96.
- Campello E, Radu CM, Duner E, et al. Activated platelet-derived and leukocyte-derived circulating microparticles and therisk of thrombosis in heparin-induced thrombocytopenia: a role for PF4-bearing microparticles. Cytometry Part B 2018; 94B: 334-341.
- Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension 2006; 48: 180-186.
- van der Zee PM, Biro E, Ko Y, et al. P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem 2006; 52: 657-664.
- Michelsen AE, Brodin E, Brosstad F, Hansen JB. Increased level of platelet microparticles in survivors of myocardial infarction. Scand J Clin Lab Invest 2008; 68: 386-392.
- Chironi GN, Simon A, Boulanger CM, et al. Circulating microparticles may influence early carotid artery remodeling. J Hypertens 2010; 28: 789-796.
- Braig D, Nero TL, Koch HG, et al. T ransitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun 2017; 8: 14188. doi:10.1038/ncomms14188
- Thiele JR, Habersberger J, Braig D, et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation 2014; 130: 35-50.
- Molins B, Pena E, de la Torre R, Badimon L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc Res 2011; 92: 328-337.
- Xu PC, Lin S, Yang XW, et al. C-reactive protein enhances activation of coagulation system and inflammatory response through dissociating into monomeric form in antineutrophil cytoplasmic antibody-associated vasculitis. BMC Immunol 2015; 16: 10. doi.org/10.1186/s12865-015-0077-0
- Thiele JR, Zeller J, Kiefer J, et al. A conformational change in C-reactive protein enhances leukocyte recruitment and reactive oxygen species generation in ischemia/reperfusion injury. Front Immunol 2018; 9: 675. doi: 10.3389/fimmu.2018.00675
- de la Torre R, Pena E, Vilahur G, Slevin M, Badimon L. Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: pentraxins and platelet deposition. J Thromb Haemost 2013; 11: 2048-2058.
- Sobol AB, Mochecka A, Selmaj K, Loba J. Is the relationship between aspirin responsivenness and clinical aspects of ischemic stroke? Adv Clin Exp Med 2009; 18: 473-479.
- Lau WC, Gurbel PA, Watkins PB, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004; 109: 166-171.
- Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357: 2001-2015.
- Aradi D, Storey RF, Komocsi A, Trenk D, Gulba D, Kiss RG. Expert position paper on the role of platelet function testing in patients undergoing percutaneous coronary intervention. Eur Heart J 2014; 35: 209-215.
- Wisniewski A. Multifactorial background for a low biological response to antiplatelet agents used in stroke prevention. Medicina 2021; 57: 59. doi.org/10.3390/medicina5701005985
- Thomas MR, Storey RF. Clinical significance of residual platelet reactivity in patients treated with platelet P2Y12 inhibitors. Vasc Pharmacol 2016; 84: 25-27.
- Winter MP, Schneeweiss T, Cremer R. Platelet reactivity patterns in patients treated with dual antiplatelet therapy. Eur J Clin Invest 2019; 49: e13102. doi: 10.1111/eci.13102
- Maree AO, Fitzgerald DJ. Variable platelet response to aspirin and clopidogrel in atherothrombotic disease. Circulation 2007; 115: 2196-2207.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Variability in individual responsiveness to clopidogrel: clinical implications, management, and future perspectives. J Am Coll Cardiol 2007; 49: 1505-1516.
- Angiolillo DJ. Variability in responsiveness to oral antiplatelet therapy. Am J Cardiol 2009; 103: 27-34.
- Gasparyan AY, Watson T, Lip GY. The role of aspirin in cardiovascular protection. J Am Coll Cardiol 2008; 51: 1829-1843.
- Tourmousoglou CE, Rokkas CK. Clopidogrel and aspirin in cardiovascular medicine: responders or not-current best available evidence. Cardiovasc Hematol Agents Med Chem 2008; 6: 312-322.
- Wisniewski A, Filipska K. The phenomenon of clopidogrel high on-treatment platelet reactivity in ischemic stroke subjects: a comprehensive review. Int J Mol Sci 2020; 21: 6408. doi:10.3390/ijms21176408
- Jastrzebska M, Lisman D, Szelepajlo A, et al. Evaluation of platelet reactivity during combined antiplatelet therapy in patients with stable coronary artery disease in relation to diabetes type 2 and the GPIIB/IIIA receptor gene polymorphism. J Physiol Pharmacol 2019; 70: 175-185.
- Xiao Z, Pierre Theroux P. Clopidogrel inhibits platelet-leukocyteinteractions and thrombin receptoragonist peptide-induced platelet activationin patients with an acute coronary syndrome. J Am Coll Cardiol 2004; 43: 1982-1988.
- Zakynthinos E, Pappa N. Inflammatory biomarkers in coronary artery disease. J Cardiol 2009; 53: 317-333.
- Sestito A, Sgueglia GA, Spinelli A, et al. Increased platelet reactivity in unstable angina patients is not related to C-reactive protein levels. Platelets 2006; 17: 336-339.
- Boncler M, Luzak B, Rozalski M, Golanski J, Rychlik B, Watala C. Acetylsalicylic acid is compounding to antiplatelet effect of C-reactive protein. Thromb Res 2007; 119: 209-216.
- Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002; 99: 3962-3970.
- Behan MW, Fox SC, Heptinstall S, Storey RF. Inhibitory effects of P2Y12 receptor antagonists on TRAP-induced platelet aggregation, procoagulant activity, microparticle formation and intracellular calcium responses in patients with acute coronary syndromes. Platelets 2005; 16: 73-80.
- Bulut D, Becker V, Mugge A. Acetylsalicylate reduces endothelial and platelet-derived microparticles in patients with coronary artery disease. Can J Physiol Pharmacol 2011; 89: 239-244.
- Kafian S, Mobarrez F, Wallen H, Samad B. Association between platelet reactivity and circulating derived microvesicles in patients with acute coronary syndrome. Platelets 2015; 26: 467-473.
- Franca CN, Pinheiro LFM, Izar MC. Endothelial progenitor cell mobilization and platelet microparticle release are influenced by clopidogrel plasma levels in stable artery disease. Circ J 2012; 76: 729-736.
- Kalantzi KI, Dimittriou AA, Goudevenos JA, Tselepsis AD. The platelets hyporesponsiveness to clopidogrel in acute coronary syndrome patients treated with 75 mg/day clopidogrel may be overcome within 1 month of treatment. Platelets 2012; 23: 121-131.
- Gasecka A, Nieuwland R, van der Pol E, et al. P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets. Cardiol J 2019; 26: 782-789.
- Jastrzebska M, Chelstowski K, Wodecka A, Siennicka A, Clark J, Nowacki P. Factors influencing multiplate whole blood impedance platelet aggregometry measurments, during aspirin treatment in acute ischemic stroke: a pilot study. Blood Coagul Fibrinolysis 2013; 24: 830-838.
- Jastrzebska J, Marcinowska Z, Oledzki S, et al. Variable gender-dependent platelet responses to combined antiplatelet therapy, in patients with stable coronary artery disease. J Physiol Pharmacol 2018; 69: 595-605.
A c c e p t e d : February 26, 2021