ORALLY ADMINISTERED ANGIOTENSIN-CONVERTING ENZYME-INHIBITORS CAPTOPRIL AND ISOLEUCINE-PROLINE-PROLINE HAVE DISTINCT EFFECTS ON LOCAL RENIN-ANGIOTENSIN SYSTEM AND CORTICOSTERONE SYNTHESIS IN DEXTRAN SULFATE SODIUM-INDUCED COLITIS IN MICE
INTRODUCTION
The roles of local renin-angiotensin system (RAS) and angiotensin-converting enzyme (ACE) inhibition in colitis have been in interest of the researchers for some years. Studies describe inflammation alleviating effects for ACE inhibitors (1-7) and knockout of RAS components (8, 9), as well as inflammation-exacerbating effects for RAS overexpression (10). We have previously shown that the proteolytic ectodomain shedding of ACE, the key enzyme of systemic RAS, is increased in the intestine in an inflammation-dependent manner (11). ACE inhibitors reduce the cleavage of angiotensin I (Ang I) to angiotensin II (Ang II), thus lowering blood pressure. Besides its blood pressure elevating effects, Ang II is pro-inflammatory and promotes fibrosis in the gut (12).
Glucocorticoids (GC) are anti-inflammatory hormones which are mainly produced in the adrenal cortex but local synthesis in various tissues is important in regulation of the organic immune systems (13-15). Intestinal GC synthesis regulates immune homeostasis and reduces intestinal permeability via tight junction protein synthesis (14). Regulation of intestinal GC synthesis differs from that of the adrenal cortex. In the intestine, pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) triggers GC synthesis by the action of the transcription factor liver receptor homolog-1 (Lrh-1), whereas in the adrenal cortex, GC synthesis is mediated by steroidogenic factor-1 (SF-1) via control of ACTH (16-18). Inflammation induces intestinal GC synthesis in animal models of colitis (18). We have previously found that Ang II, the product of ACE, increases GC synthesis in vitro and that ACE protein shedding is increased by inflammation in small intestine (11). Therefore, we explored the effects of ACE inhibition on intestinal RAS and GC synthesis components in a dextran sulfate sodium (DSS) model of colitis to further elucidate their possible interactions in inflammatory conditions.
We chose to employ a widely used pharmacological ACE inhibitor captopril along with a nutritional compound, the milk casein-derived bioactive tripeptide isoleucine-proline-proline (Ile-Pro-Pro). Captopril was the first pharmacological ACE inhibitor in clinical use with antihypertensive and anti-inflammatory properties (19). Ile-Pro-Pro is a weak competitive ACE inhibitor with IC50 value of 5 mM, whereas the IC50 value of captopril is 21 nM (20). Clinical studies and animal models have demonstrated the beneficial effects of Ile-Pro-Pro on elevated blood pressure and vascular function (21-25). Proline-rich tripeptides like Ile-Pro-Pro are not well absorbed and are resistant to digestive peptidases and should therefore reach the colon lumen in some extent (26, 27).
MATERIALS AND METHODS
Animals
The study was approved by National Animal Experimentation Committee of Finland (ESAVI/114/04.10.07/2015) according to EC Directive 86/609/ECC and Finnish Experimental Animal Act 62/2006. A total of 32 Balb/c male mice were obtained from a commercial breeding company (Scanbur, Sollentuna, Sweden) at the age of eight weeks. The mice were housed in social groups of two mice per cage in specific-pathogen free laboratory conditions with 12-h light/dark cycle with room temperature of 22°C ± 2°C and relative humidity of 55% ± 15%. The mice were allowed to acclimatize for seven days before the commencement of the study. All the animals were given standard rodent feed, 2018 Teklad Global 18% Protein Rodent Diet (Harlan Laboratories, Indianapolis, IN, USA) and test drinks ad libitum. The weights of the animals were monitored daily.
Colitis model
Mice were randomly assigned to four experimental groups of eight mice each. Healthy control mice were given tap water throughout the experiment. One group was given 15.7 mg/l captopril (PHR1307-1G, Sigma-Aldrich, St. Louis, MO, USA) and one 833 mg/l Ile-Pro-Pro (H-4632, Bachem, Bubendorf, Switzerland) in their test drinks for a total of 12 days, corresponding to 0.11 mg/day of captopril and 5 mg/day of Ile-Pro-Pro based on the monitored consumption of test drinks. After 5 days, 3% dextran sodium sulfate (DSS) (TdB Concultancy, Uppsala, Sweden) was added to the drinking water of the groups receiving captopril or Ile-Pro-Pro and DSS control group for seven days, after which the animals were sacrificed. The dose of captopril was chosen based on previous studies in which captopril has been used in mouse colitis models, and studies in which captopril has been administered in drinking water (1, 28, 29). The dose of Ile-Pro-Pro was set so that its ACE-inhibiting capacity could approach that of captopril.
Assessment of macroscopic inflammation
Mice were sacrificed by cardiac puncture under isoflurane (Vetflurane 1000 mg/g, Virbac, Suffolk, UK) anesthesia. Upon sacrifice, the colons of mice were excised and their lengths measured. Macroscopic evaluation of inflammation was done as follows. The colon was opened and stool consistency scored on a scale of 0 – 2 where 0 indicates normal stool and 2 indicates watery stool. The presence of blood was scored on a scale of 0 – 2 where 0 indicates none present and 2 indicates considerable amount of visible blood The colons were cleaned and edema scored similarly on scale 0 – 2. The scores were combined to a total macroscopic score (scale 0 – 6).
Ex-vivo incubation samples
Pieces of proximal colon were incubated in pre-oxygenated Krebs buffer (119 mmol/l NaCl, 25 mmol/l NaHCO3, 15 mmol/l KCl, 11 mmol/l glucose, 1.6 mmol/l CaCl2, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4), for which the chemicals were purchased from Sigma-Aldrich, for 90 min at 37°C in gentle agitation. The samples were centrifuged at 13,300 g for 3 min. ACE (#DY1513 R&D System, Minnesota, MO, USA) and corticosterone (#500655 Cayman Chemical, Michigan, MI, USA) were measured from the supernatants and total protein (Pierce™ BCA Protein Assay Kit, Thermo Scientific) from the tissue homogenates.
Cytokine analyses
Tissue pieces of colon were homogenized with Precellys 24 homogenator (Bertin Technologies, Montigny le Bretonneux, France) in 100 mM Tris - 120 mM NaCl - 1 × protease inhibitors (#88665 Pierce Protease Inhibitor Mini Tablets, ThermoFisher Scientific, Waltham, MA USA). IL-1β, TNF-α and IL-6 concentrations were measured using AlphaLisa (AL503, AL504 and AL505, Perkin Elmer, Waltham, MA, USA) and normalized to total protein of the tissue piece.
Histological analyses
Samples of distal colon were fixed in 10% neutral-buffered formalin (Sigma-Aldrich, St. Louis, MO, USA) and embedded in paraffin blocks. Paraffin-embedded tissue slides were stained with hematoxylin and eosin dye and assessed for inflammation. Histological analysis was done according to Erben et al., (30). Briefly, inflammatory cell infiltration and epithelial erosion were each scored on a scale of 0 – 3 and summed yielding a maximum total score of 6.
Reverse transcriptase quantitative PCR
RNA was extracted from tissues using NucleoSpin RNA kit (#740955, Macherey Nagel, Duren, Germany). cDNA was transcribed using iScript™ cDNA Synthesis Kit (#1708891, BioRad, Hercules, CA, USA). RT-qPCR was run using LightCycler® 480 SYBR Green I Master (#04707516001, Roche Diagnostics Corp., Indianapolis, IN, USA) Primers for target genes were ordered from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). The primer sequences are displayed in Table 1. Target gene expression was normalized to expression of β-actin and S18 and normalization factors calculated according to the method described elsewhere (31). RT-qPCR was run using the LightCycler® 480 system.

Statistical analyses
Data were analyzed using Shapiro-Wilk test for normality and Levene’s test for homogeneity of variances. Outliers were removed in SPSS Statistics (IBM, Armonk, NY, USA). Differences between two groups were analyzed with 2-sided Student’s t-test or Mann-Whitney U test and multiple groups were analyzed with one-way ANOVA in SPSS version 22 except for the differences in weight change, which were analyzed by repeated measures ANOVA in GraphPad Prism version 5 (GraphPad Software, La Jolla, CA, USA). Correlations were calculated as Spearman’s correlations. P-values smaller than 0.05 were considered statistically significant. Data in boxplots are presented as median, min and max values and 25th and 75th percentiles and in text and tables as mean ± S.E.M.
RESULTS
Induction of colitis
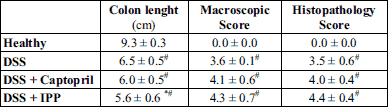
During the initial ACE-inhibitor administration period there were no differences in weight development between the study groups (data not shown). Weight of the mice in all three DSS-receiving groups began to decline after six days of DSS administration and the decline was more prominent in the group receiving captopril (Fig. 1). Histological analysis showed marked differences (P < 0.001) between healthy controls and all colitis groups (Fig. 2, Table 2). All DSS groups displayed mucosal and submucosal inflammatory cell infiltration and considerable ulceration. The histopathology scores were similar between colitis groups. The macroscopic score (Table 2) was higher (P = 0.001) and colon lengths were shorter (P < 0.001) in all colitis groups compared to healthy controls. The colon shortening was more prominent in mice receiving Ile-Pro-Pro (Table 2, P = 0.044) compared to the DSS controls without differences in macroscopic or histopathology scores.
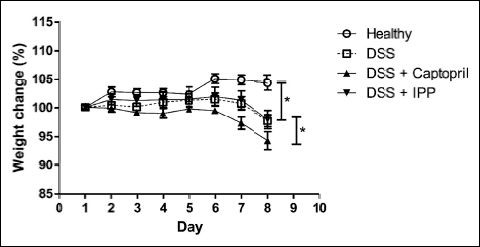 |
Fig. 1. Weight development from the beginning of DSS administration. The weights of DSS control mice were significantly lower than those of healthy controls. Captopril treatment caused a further reduction in weight compared to DSS controls. * denotes P < 0.05. IPP, Ile-Pro-Pro. |
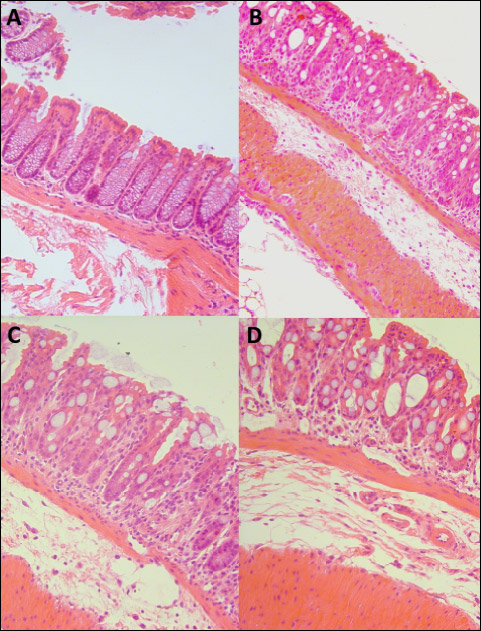 |
Fig. 2. H&E stained sections of (A) Healthy (B) DSS (C) DSS + Captopril and (D) DSS + IPP group colons. Objective magnifications × 20. All DSS group colons displayed considerable distortions of the crypt structures, inflammatory cell infiltration and submucosal edema. IPP, Ile-Pro-Pro. |
* denotes P < 0.05 compared to DSS group. # denotes P < 0.05 compared to healthy controls. IPP, Ile-Pro-Pro.
Angiotensin-converting enzyme shedding is reduced by Ile-Pro-Pro
ACE shedding, measured as ACE protein released into the incubation fluid, was similar between healthy mice and DSS controls (Fig. 3). However, Ile-Pro-Pro administration reduced ACE shedding from the colon tissue to 0.58-fold compared to DSS controls (P = 0.014). There was a less prominent reduction of ACE shedding by captopril to 0.77-fold which was not statistically significant (P = 0.245). The ACE protein concentrations were 10.3 ± 0.6 ng/mg of protein for healthy mice, 9.2 ± 0.9 ng/mg for DSS controls, 7.1 ± 0.8 ng/mg for captopril and 5.4 ± 0.8 ng/mg for Ile-Pro-Pro group.
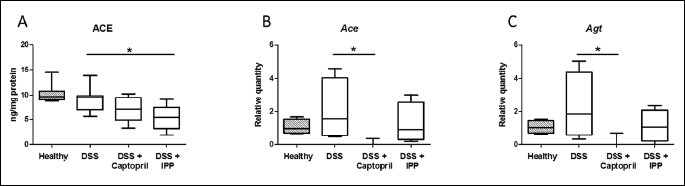
* denotes P < 0.05. IPP, Ile-Pro-Pro.
Colitis-induced corticosterone production is not influenced by angiotensin-converting enzyme inhibition
DSS increased corticosterone production 1.97-fold in the proximal colon compared to healthy mice (P < 0.001) (Fig. 4). Corticosterone production was 0.73-fold in the captopril and 0.89-fold in the Ile-Pro-Pro group of the DSS controls and not statistically significant.
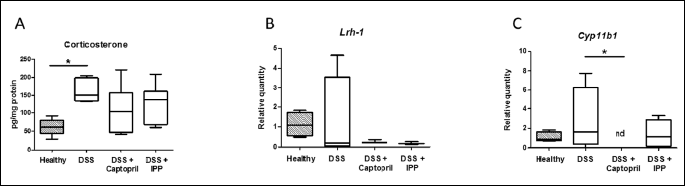
Expression of angiotensin-converting enzyme, angiotensinogen and Cyp11b1 are reduced in mice receiving captopril
To test whether reduction of ACE shedding was associated with decrease in Ace expression by Ile-Pro-Pro, the relative quantities of Ace mRNA were measured. Angiotensinogen (Agt), Cyp11b1 and Lrh-1 mRNA levels were also quantified. DSS alone did not significantly affect the expression of these genes compared to healthy mice (Figs. 3 and 4). The relative expression in DSS controls compared to healthy controls were 1.40-fold for Ace, 1.53-fold for Agt, 0.56-fold for Lrh-1 and 2.75-fold for Cyp11b1, which was highly variable in the DSS control group. Ace expression in Ile-Pro-Pro group was 0.60-fold of DSS controls but not significantly lower (Fig. 3A). Ace mRNA expression did not correlate with ACE protein shedding (rp = 0.231, P = 0.407). Captopril lowered Ace mRNA expression to 0.17-fold (P = 0.025) and Agt to 0.18-fold (P = 0.030) of DSS controls (Fig. 3B, 3C and 4C). Cyp11b1 was not detected in captopril-treated colons (P = 0.040 compared to DSS controls). Lrh-1 expression was not affected by the treatments. Ile-Pro-Pro had no effect in mRNA expression of the studied genes which were expressed at levels of 0.59-fold for Ace, 0.41-fold for Agt, 0.33-fold for Lrh-1 and 0.55-fold for Cyp11b1 compared to DSS controls.
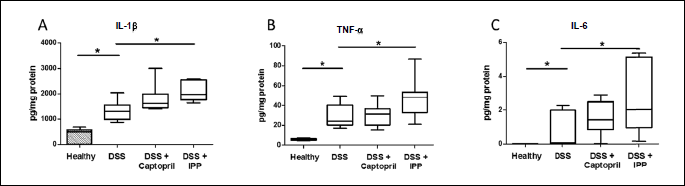
Ile-Pro-Pro increases pro-inflammatory cytokine production in the inflamed colon
IL-1β (P < 0.001), TNF-α (P = 0.009) and IL-6 (P = 0.004) were significantly increased in the colons of DSS controls compared to healthy mice (Fig. 5). TNF-α (P = 0.046), IL-1β (P = 0.006) and IL-6 (P = 0.030) were further increased in mice receiving Ile-Pro-Pro but not captopril. TNF-α concentrations were 6.0 ± 0.4 pg/mg of protein in healthy controls, 28.6 ± 4.1 pg/mg in the DSS controls, 30.4 ± 3.7 pg/mg in the captopril group and 47.6 ± 6.5 pg/mg in the Ile-Pro-Pro group. IL-1β concentrations were 351 ± 98 pg/mg in the healthy controls, 1318 ± 137 pg/mg in the DSS controls, 1803 ± 176 in the captopril group and 2091 ± 131 pg/mg in the Ile-Pro-Pro group. IL-6 was not detected in healthy controls and the concentrations were 0.8 ± 0.4 pg/mg in the DSS control group, 1.5 ± 0.3 in the captopril group and 2.8 ± 0.9 pg/mg in the Ile-Pro-Pro group.
DISCUSSION
There has been a growing interest in intestinal RAS as a regulator of inflammation. ACE-inhibition has previously been reported to impede experimental colitis (1, 2, 4). Different research groups have reported that intraperitoneal administration of captopril and lisinopril, oral administration of enalapril, rectal administration of enalaprilat or oral and subcutaneous dosing of losartan improve colitis in various colitis models in wildtype and genetically predisposed mice and rats (1, 2, 4-8). Furthermore, angiotensin receptor subtype 1 (AT1R) knockout mice are resistant to DSS colitis (3) and renin over-expression exacerbates 2, 4, 6-trinitrobenzenesulfonic acid -induced colitis (10). The so called alternative RAS components make this entity even more interesting. Angiotensin(1-7), the breakdown product of angiotensin II, and activation of its target receptor, the Mas receptor, can accelerate mucosal healing as shown in experimental model of gastric ulcers (32).
The present study further elucidates the consequences of ACE inhibition on local RAS and regulation of glucocorticoid synthesis in experimental colitis. For that purpose, we employed a pharmacological ACE inhibitor captopril and a nutritional ACE-inhibiting tripeptide Ile-Pro-Pro given orally in drinking fluid in a DSS model of colitis.
Although the studies mentioned above have found ACE inhibition and angiotensin receptor blockade to alleviate colitis, in our hands orally administered ACE inhibition did not improve the colitis outcome as measured by weight loss, changes in histopathology or biochemical inflammatory markers. All DSS groups developed marked colitis based on weight change, microscopic analyses of inflammation and increased production of pro-inflammatory cytokines IL-1β, TNF-α and IL-6, and the anti-inflammatory glucocorticoid hormone corticosterone. However, the tripeptide Ile-Pro-Pro, which has beneficial effects on blood pressure and vascular function (21-25), further exacerbated DSS colitis. Compared to the DSS control group, Ile-Pro-Pro group exhibited higher colonic IL-1β and TNF-α concentrations, shorter colon lengths and a trend for higher macroscopic and histopathology scores. In contrast, although the body weights of the mice in the captopril group were lower than the DSS control mice, we did not observe any significant differences in inflammatory markers, macroscopic, and histological scores between these groups. However, captopril administration reduced the colonic gene expression of Ace, Agt, and Cyp11b1, the rate-limiting enzyme in corticosterone synthesis, indicating captopril-induced effects on the local RAS and steroidogenesis. Ile-Pro-Pro had no effect on colonic gene expression of the studied genes suggesting distinctive mechanistic differences between these two ACE-inhibiting agents.
As to why there was no improvement of colitis by captopril in this study, we speculate that a lower dose of ACE inhibitors and the diffuse area of effect after oral administration were not sufficient enough to prevent colitis. Most studies done with ACE inhibitors have employed other than oral administration routes. However, captopril has a short half-life and therefore oral administration in drinking water allows a more even intake of captopril during the day. On the other hand, captopril has a distinct sulfuric smell and taste, which might have reduced the daily amount of fluid and food intake and contributed to lower bodyweight of the animals in the captopril group. Similar observations of lower bodyweight gain have been made in other studies using captopril (33).
Our results indicate that Ile-Pro-Pro has a pro-inflammatory effect on colitis as exhibited by reduced colon length and increased colonic levels of pro-inflammatory cytokines. This finding is in interesting considering that no studies have found adverse effects even with high doses of Ile-Pro-Pro (34-36). In this study, the animals tolerated the given dose of Ile-Pro-Pro well during the initial Ile-Pro-Pro administration phase and no adverse effects or phenotypic changes were observed before beginning of DSS administration. We used a high dose of Ile-Pro-Pro to reach a similar order of ACE inhibition as with captopril. Therefore, some of Ile-Pro-Pro’s observed effects could be due to ACE inhibition. However, highlighted by the distinct effects of Ile-Pro-Pro and captopril on colonic RAS mRNA expression, not all observed effects of Ile-Pro-Pro can be due to ACE inhibition only. Although Ile-Pro-Pro has the strongest affection to ACE, it also activates cathepsin G and inhibits prolyl oligopeptidase activity (37, 38). Cathepsins are peptidases of which several are of importance in intestinal inflammation (39). Cathepsin B, D, G and L are increased in colitis ulcerosa patients and their respective inhibitors alleviate inflammation in animal models (40, 41). Cathepsin G cleaves angiotensinogen and Ang I to Ang II (42). On the other hand cathepsin G inactivates IL-6 and TNF-α (39). It could be speculated that the high concentration of Ile-Pro-Pro in the inflamed colon could drive inflammation via cathepsins e.g. cathepsin G.
The mechanisms and underlying function of ACE shedding in vasculature and intestine are largely unknown. Similarly, we can only speculate how Ile-Pro-Pro reduces ACE shedding in the intestine. Disintegrin and metalloproteinase domain-containing protein 9 (ADAM9) has been identified as one of the enzymes which cleave, or shed, ACE from cell membranes (43). ACE shedding leading to increase in circulating ACE occurs in some diseases like Gaucher’s disease, sarcoidosis, a systemic inflammatory disease with unknown aetiology, diabetes, and due to some ACE polymorphisms (44, 45). We have previously demonstrated that ACE is shed from both small and large intestine following inflammation in mice (11). Treating pulmonary microvascular endothelial cells with LPS to simulate sepsis leads to increased ACE shedding and reduced mRNA expression of Ace (43, 46). The lack of correlation between ACE protein shedding and Ace mRNA expression suggests that ACE shedding is regulated at protein level and not simply dependent on ACE production. This claim is supported by our previous studies showing that ACE shedding is not dependent on tissue level of ACE (11). ACE shedding and biosynthesis are increased in the lung by captopril and enalapril (47, 48). Our results of decreased ACE shedding in the colon are in contrast with increased pulmonary endothelial shedding of ACE in response to ACE inhibitors and highlights the difference in regulation of local and systemic RAS. Interestingly, ACE shedding seems to response analogously to inflammation in vasculature and intestine, although the response to ACE inhibition is different.
Ang II stimulates glucocorticoid secretion in adrenal glands. Previously we have seen an increase in corticosterone secretion in Ang II-treated small intestine samples (11). Therefore we wanted to test, whether ACE inhibition would reduce DSS-induced corticosterone production. Captopril lowered colonic Cyp11b1 mRNA expression but the DSS-induced corticosterone production in colon was not modulated by ACE inhibition in vivo. The effect of ACE inhibition by captopril on Cyp11b1 mRNA expression in colon is a novel finding, which gives further evidence to a hypothesis that intestinal RAS, in part, regulates local intestinal glucocorticoid production. Aldosterone levels were not measured in this study, but it is worth noting that Cyp11b1 enzyme functions in aldosterone synthesis pathway as well. The effect of ACE inhibition on aldosterone synthesis in the intestine is virtually an unexplored topic, which would be a lucrative topic for future studies. Cyp11b1 mRNA expression is not affected by captopril in either the zona glomerulosa, which produces aldosterone, or the zona fasciculata, which produces glucocorticoids, of the adrenal glands (49) or by AT1R blockade by losartan in lung, heart or kidney (50). Therefore, captopril-mediated ACE inhibition seems to differentially regulate steroidogenic Cyp11b1 expression in colon and adrenals, which should be verified in further studies.
Taken together, this study highlights the distinct effects of two ACE inhibiting agents, captopril and the nutritional bioactive peptide Ile-Pro-Pro, on intestinal renin-angiotensin system and adds evidence of the crosstalk between intestinal RAS and glucocorticoid synthesis. Furthermore, this study has revealed putative evidence of tissue-specific actions of captopril on intestinal RAS and Cyp11b1 expression.
Abbreviations: ACE, angiotensin-converting enzyme; ADAM9, disintegrin and metalloproteinase domain-containing protein 9; Agt, angiotensinogen; Ang II, angiotensin II; AT1R, angiotensin receptor subtype 1; DSS, dextran sodium sulfate; GC, glucocorticoid; Ile-Pro-Pro, isoleucine-proline-proline; Lrh-1, liver receptor homolog-1; RAS, renin-angiotensin system.
Author contributions: H. Salmenkari, H. Vapaatalo and R. Korpela designed the study plan; H. Salmenkari, R. Forsgard and M. Holappa performed the experiments; H. Salmenkari analyzed the data; H. Salmenkari, H. Vapaatalo and R. Korpela wrote the paper.
Acknowledgements: We thank Niina Siiskonen at Tissue Preparation and Histochemistry Unit, Medicum, Depatment of Anatomy, University of Helsinki for preparation of the H&E stained tissue slides.
Supportive foundations: Foundation for Clinical Chemistry Research, Finland and Finska Lakaresallskapet and Einar och Karin Stroems Stiftelse, Finland.
Conflict of interests: None declared.
REFERENCES
- Jahovic N, Ercan F, Gedik N, Yuksel M, Sener G, Alican I. The effect of angiotensin-converting enzyme inhibitors on experimental colitis in rats. Regul Pept 2005; 130: 67-74.
- Spencer AU, Yang H, Haxhija EQ, Wildhaber BE, Greenson JK, Teitelbaum DH. Reduced severity of a mouse colitis model with angiotensin converting enzyme inhibition. Dig Dis Sci 2007; 52: 1060-1070.
- Mizushima T, Sasaki M, Ando T, et al. Blockage of angiotensin II type 1 receptor regulates TNF-αlpha-induced MAdCAM-1 expression via inhibition of NF-kappaB translocation to the nucleus and ameliorates colitis. Am J Physiol Gastrointest Liver Physiol 2010; 298: G255-G266.
- Koga H, Yang H, Adler J, Zimmermann EM, Teitelbaum DH. Transanal delivery of angiotensin converting enzyme inhibitor prevents colonic fibrosis in a mouse colitis model: development of a unique mode of treatment. Surgery 2008; 144: 259-268.
- Lee C, Chun J, Hwang SW, Kang SJ, Im JP, Kim JS. Enalapril inhibits nuclear factor-kappaB signaling in intestinal epithelial cells and peritoneal macrophages and attenuates experimental colitis in mice. Life Sci 2014; 95: 29-39.
- Wengrower D, Zanninelli G, Pappo O, et al. Prevention of fibrosis in experimental colitis by captopril: the role of tgf-beta1. Inflamm Bowel Dis 2004; 10: 536-545.
- Sueyoshi R, Ignatoski KM, Daignault S, Okawada M, Teitelbaum DH. Angiotensin converting enzyme-inhibitor reduces colitis severity in an IL-10 knockout model. Dig Dis Sci 2013; 58: 3165-3177.
- Inokuchi Y, Morohashi T, Kawana I, Nagashima Y, Kihara M, Umemura S. Amelioration of 2,4,6-trinitrobenzene sulphonic acid induced colitis in angiotensinogen gene knockout mice. Gut 2005; 54: 349-356.
- Katada K, Yoshida N, Suzuki T, et al. Dextran sulfate sodium-induced acute colonic inflammation in angiotensin II type 1a receptor deficient mice. Inflamm Res 2008; 57: 84-91.
- Shi Y, Liu T, He L, et al. Activation of the renin-angiotensin system promotes colitis development. Sci Rep 2016; 6: 27552. doi: 10.1038/srep27552.
- Salmenkari H, Issakainen T, Vapaatalo H, Korpela R. Local corticosterone production and angiotensin-I converting enzyme shedding in a mouse model of intestinal inflammation. World J Gastroenterol 2015; 21: 10072-10079.
- Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther 2012; 35: 414-428.
- Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am J Physiol Gastrointest Liver Physiol 2007; 292: G590-G598.
- Kostadinova F, Schwaderer J, Sebeo V, Brunner T. Why does the gut synthesize glucocorticoids? Ann Med 2014; 46: 490-497.
- Cima I, Corazza N, Dick B, et al. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med 2004; 200: 1635-1646.
- Mueller M, Cima I, Noti M, et al. The nuclear receptor Lrh-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med 2006; 203: 2057-2062.
- Noti M, Corazza N, Tuffin G, Schoonjans K, Brunner T. Lipopolysaccharide induces intestinal glucocorticoid synthesis in a TNF-αlpha-dependent manner. FASEB J 2010; 24: 1340-1346.
- Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010; 207: 1057-1066.
- El Desoky ES. Drug therapy of heart failure: an immunologic view. Am J Ther 2011; 18: 416-425.
- Nakamura Y, Yamamoto N, Sakai K, Okubo A, Yamazaki S, Takano T. Purification and characterization of angiotensin I-converting enzyme inhibitors from sour milk. J Dairy Sci 1995; 78: 777-783.
- Turpeinen AM, Ehlers PI, Kivimaki AS, et al. Ile-Pro-Pro and Val-Pro-Pro tripeptide-containing milk product has acute blood pressure lowering effects in mildly hypertensive subjects. Clin Exp Hypertens 2011; 33: 388-396.
- Jakala P. Milk protein-derived bioactive tripeptides Ile-Pro-Pro and Val-Pro-Pro protect endothelial function in vitro in hypertensive rats. J Funct Foods 2009; 1: 226-273.
- Ehlers PI. High blood pressure-lowering and vasoprotective effects of milk products in experimental hypertension. Br J Nutr 2011; 106: 1353-1363.
- Turpeinen AM, Jarvenpaa S, Kautiainen H, Korpela R, Vapaatalo H. Antihypertensive effects of bioactive tripeptides-a random effects meta-analysis. Ann Med 2013; 45: 51-56.
- Nakamura T, Mizutani J, Ohki K, et al. Casein hydrolysate containing Val-Pro-Pro and Ile-Pro-Pro improves central blood pressure and arterial stiffness in hypertensive subjects: a randomized, double-blind, placebo-controlled trial. Atherosclerosis 2011; 219: 298-303.
- Ohsawa K, Satsu H, Ohki K, Enjoh M, Takano T, Shimizu M. Producibility and digestibility of antihypertensive beta-casein tripeptides, Val-Pro-Pro and Ile-Pro-Pro, in the gastrointestinal tract: analyses using an in vitro model of mammalian gastrointestinal digestion. J Agric Food Chem 2008; 56: 854-858.
- Ten Have GA, van der Pijl PC, Kies AK, Deutz NE. Enhanced lacto-tri-peptide bio-availability by co-ingestion of macronutrients. PLoS One 2015; 10: e0130638. doi: 10.1371/journal.pone.0130638
- Godsel LM, Leon JS, Wang K, Fornek JL, Molteni A, Engman DM. Captopril prevents experimental autoimmune myocarditis. J Immunol 2003; 171: 346-3 52.
- Leon JS, Wang K, Engman DM. Captopril ameliorates myocarditis in acute experimental Chagas disease. Circulation 2003; 107: 2264-2269.
- Erben U, Loddenkemper C, Doerfel K, et al. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol 2014; 7: 4557-4576.
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007; 8: R19.
- Pawlik MW, Kwiecien S, Ptak-Belowska A, et al. The renin-angiotensin system and its vasoactive metabolite angiotensin-(1-7) in the mechanism of the healing of preexisting gastric ulcers. The involvement of Mas receptors, nitric oxide, prostaglandins and proinflammatory cytokines. J Physiol Pharmacol 2016; 67: 75-91.
- Aziriova S, Repova K, Krajcirovicova K, et al. Effect of ivabradine, captopril and melatonin on the behaviour of rats in L-nitro-arginine methyl ester-induced hypertension. J Physiol Pharmacol 2016; 67: 895-902.
- Nonaka A, Nakamura T, Hirota T, et al. The milk-derived peptides Val-Pro-Pro and Ile-Pro-Pro attenuate arterial dysfunction in L-NAME-treated rats. Hypertens Res 2014; 37: 703-707.
- Gleeson JP, Heade J, Ryan SM, Brayden DJ. Stability, toxicity and intestinal permeation enhancement of two food-derived antihypertensive tripeptides, Ile-Pro-Pro and Leu-Lys-Pro. Peptides 2015; 71: 1-7.
- Ponstein-Simarro Doorten AY, vd Wiel JA, Jonker D. Safety evaluation of an IPP tripeptide-containing milk protein hydrolysate. Food Chem Toxicol 2009; 47: 55-61.
- Siltari A, Kivimaki AS, Ehlers PI, Korpela R, Vapaatalo H. Effects of milk casein derived tripeptides on endothelial enzymes in vitro; a study with synthetic tripeptides. Arzneimittelforschung 2012; 62: 477-4781.
- Lehtinen R, Jauhiainen T, Kankuri E, et al. Effects of milk casein-derived tripeptides Ile-Pro-Pro, Val-Pro-Pro, and Leu-Pro-Pro on enzymes processing vasoactive precursors in vitro. Arzneimittelforschung 2010; 60: 182-185.
- Vergnolle N. Protease inhibition as new therapeutic strategy for GI diseases. Gut 2016; 65: 1215-1224.
- Menzel K, Hausmann M, Obermeier F, et al. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin Exp Immunol 2006; 146: 169-180.
- Dabek M, Ferrier L, Annahazi A, et al. Intracolonic infusion of fecal supernatants from ulcerative colitis patients triggers altered permeability and inflammation in mice: role of cathepsin G and protease-activated receptor-4. Inflamm Bowel Dis 2011; 17: 1409-1414.
- Holappa M, Vapaatalo H, Vaajanen A. Ocular renin-angiotensin system with special reference in the anterior part of the eye. World J Ophthalmol 2015; 5: 110-124.
- English WR, Corvol P, Murphy G. LPS activates ADAM9 dependent shedding of ACE from endothelial cells. Biochem Biophys Res Commun 2012; 421: 70-75.
- Ehlers MR, Gordon K, Schwager SL, Sturrock ED. Shedding the load of hypertension: the proteolytic processing of angiotensin-converting enzyme. S Afr Med J 2012; 102: 461-464.
- Oh JK, Han IK, Kim JW, et al. The changes of serum-ACE, plasma renin activity and aldosterone in the diabetics with hypertension. Korean J Intern Med 1986; 1: 26-30.
- Hermanns MI, Muller AM, Tsokos M, Kirkpatrick CJ. LPS-induced effects on angiotensin I-converting enzyme expression and shedding in human pulmonary microvascular endothelial cells. in vitro Cell Dev Biol Anim 2014; 50: 287-295.
- Fyhrquist F, Forslund T, Tikkanen I, Gronhagen-Riska C. Induction of angiotensin I-converting enzyme rat lung with Captopril (SQ 14225). Eur J Pharmacol 1980; 67: 473-475.
- Forslund T, Fyhrquist F, Gronhagen-Riska C, Tikkanen I. Induction of angiotensin-converting enzyme with the ACE inhibitory compound MK-421 in rat lung. Eur J Pharmacol 1982; 80: 121-125.
- LeHoux JG, Tremblay A. in vivo regulation of gene expression of enzymes controlling aldosterone synthesis in rat adrenal. J Steroid Biochem Mol Biol 1992; 43: 837-846.
- Metsarinne KP, Helin KH, Saijonmaa O, Stewen P, Sirvio ML, Fyhrquist FY. Tissue-specific regulation of angiotensin-converting enzyme by angiotensin II and losartan in the rat. Blood Press 1996; 5: 363-370.
A c c e p t e d : May 26, 2017