HISTOPATHOLOGICAL PARAMETERS OF THE SPINAL CORD IN DIFFERENT PHASES OF EXPERIMENTAL AUTOIMMUNE ENCEPHALOMYELITIS. A MOUSE MODEL OF MULTIPLE SCLEROSIS EXAMINED BY CLASSICAL STAININGS COMBINED WITH IMMUNOHISTOCHEMISTRY
2Medical Departament, Novartis Poland Sp. z o.o., Warsaw, Poland
INTRODUCTION
Experimental autoimmune encephalomyelitis (EAE) is an autoimmune disease of the central nervous system (CNS) and a commonly used animal model of multiple sclerosis (MS) that can be induced in mice by immunization with myelin oligodendrocyte glycoprotein (MOG) as the autoreactive antigen (1, 2). The breakdown of the blood-brain barrier (BBB), leukocyte infiltration into the nerve tissue and proinflammatory cytokine production by the infiltrating cells are the main pathomechanisms responsible for the development of EAE/MS (1).
Inflammation, neurodegeneration and axonal damage are the most important pathological processes in MS and EAE (2, 3). Demyelination underlies sensorimotor symptoms that predominate in the clinical profile of EAE (4).
Due to the limited availability of human CNS samples, EAE is frequently used in mechanism-oriented studies (5). Hematoxylin/eosin (HE), Luxol Fast Blue (LFB) and Bielschowsky silver impregnation (BSI) are the basic histological staining methods used in assessment of inflammation, demyelination and axonal injury in CNS. This type of histopathological analysis visualizes possible histological changes and is useful for monitoring progression and course of EAE (6-8).
The aim of this study was to quantify the neuropathological parameters of MS/EAE: inflammatory infiltration, demyelination, and axonal injury in the successive phases of EAE: onset, peak and chronic, using histological images of spinal cord samples. We hypothesize that the advancement of these pathological processes is parallel to the progression of clinical symptoms and therefore histological assessment of these parameters in the spinal cord can be helpful in evaluation of EAE severity and effectiveness of therapies.
MATERIALS AND METHODS
Animals
Pathogen-free C57BL/6 mice (female, 10–11 weeks old, weight 19–24 g) were purchased from the Center for Experimental Medicine of Bialystok Medical University, Poland (strain imported from Jackson Laboratory). Mice were housed, five per cage, in animal house of the Jagiellonian Centre for Experimental Therapeutics (JCET), Cracow, under 12-hour light-dark cycle in temperature-controlled environment (22±2°C, 55±10% humidity). Standard irradiated laboratory chow and water were available ad libitum. All experiments were conducted in compliance with the Council Directive 2010/63EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes and approved by the First and the Second Local Ethics Committees in Cracow, Poland (Permissions 118/2015 and 274/2018).
Induction of experimental autoimmune encephalomyelitis
For induction of EAE (Fig. 1b), on day 0 mice were immunized with 200 µL injection of Hooke Kits™ EAE emulsion (Hooke Laboratories, Lawrence, MA, USA) containing MOG35–55 peptide emulsified in Complete Freund’s Adjuvant (CFA) including 4 mg/mL heat-killed Mycobacterium tuberculosis (H37Ra). Emulsion was administered subcutaneously at two sites (100 µl behind the neck and 100 µl in hind flank). On day 0 and day 1, each mouse was also injected intraperitoneally (i.p.) with 340 µl of Bordatella pertussis pertussis toxin (PTx) dissolved in phosphate-buffered saline (PBS) 2 h after the administration of the emulsion and again 24 h later. Control mice were injected with CFA and PTx dissolved in PBS (Hooke Control Kits™, Hooke Laboratiores, USA) according to the same schedule (Fig. 1b).
Monitoring of experimental autoimmune encephalomyelitis course
The clinical symptoms of EAE were scored following protocols provided by the Hooke Laboratories and as reported previously (8-11). The control mice did not show any symptoms of the disease. The clinical behavior of mice was scored daily (from 0 to 30 days) following protocols provided by the Hooke Laboratories (Fig. 1a). Disease severity of EAE was evaluated using a 0–3 scale: (0) no clinical disease; 0.5 - limp tip of tail; 1 - limp tail; 1.5 - limp tail and hind leg inhibition; 2 - limp tail and weakness of hind legs; 2.5 - limp tail and dragging of hind legs; 3 - limp tail and complete paralysis of hind legs (0.5 gradations represent intermediate scores).
Spinal cord collection
To collect spinal cords, mice were anaesthetized with ketamine/xylazine cocktail (100 mg/kg and 10 mg/kg, respectively, through intraperitoneal injection). Mice were perfused transcardially with ice-cold PBS for 5 minutes, followed by 4% paraformaldehyde for 10 minutes. The spinal cords were removed and fixed in buffered 4% paraformaldehyde at 4°C for 4 hours. After overnight incubation in 5% sucrose solution in PBS at 4°C, they were flash-frozen in optimal cutting temperature compound (OCT, Shandon Cryomatrix; ThermoFisher Sci., Waltham, MA, USA) at –80°C. Eight µm thick serial cryosections were collected on poly-L-lysine coated slides and air-dried. The area of the spinal cord examined included the lumbar region, an area frequently and rapidly involved in EAE.
Experimental groups and time-dependent spinal cord collection
According to the experimental schedule presented in our previous publications (8-11), the immunized mice (n=15) were sacrificed at different time points (Fig. 1a, 1b), representing three successive phases of the disease: onset (day 13; n=5), peak (day 18; n=5) and chronic (day 30; n=5). Control non-immunized mice (n=15) were sacrificed on days corresponding to previously mentioned phases (n=5 per phase).
Histopathology and immunofluorescence staining
Three staining methods were applied: hematoxylin and eosin (HE) to analyze the degree of inflammatory infiltration, Luxol Fast Blue (LFB) to visualize demyelinated areas and Bielschowsky silver impregnation (BSI) to detect axonal damage.
In HE method, sections were stained with hematoxylin ready-to-use (DiaPath, Martinengo, Italy) for 10 min. and then with 1% yellow eosin (Chempur, Piekary Slaskie, Poland) in distilled water for 3 min.
In LFB method, sections were stained with 0.1% Luxol Fast Blue (Sigma-Aldrich, St. Louis, MO, USA) solution in acetic alcohol at 56°C overnight, next they were differentiated in 0.05% lithium carbonate and counterstained in 0.1% Cresyl Fast Violet (Sigma-Aldrich, St. Louis, MO, USA) aqueous solution.
In BSI method, the nerve fibers were pretreated with 20% silver nitrate (Sigma-Aldrich, St. Louis, MO, USA) for 15 min, then treated with silver nitrate solution in ammoniacal water (concentrated ammonium hydroxide was added drop by drop to make solution cloudy) for 30 min at 40°C, and next developed in developer working solution for 1 min (made fresh: 8 drops of developer stock solution: a mixture of formaldehyde, citric acid and nitric acid in distilled water, 8 drops of concentrated ammonium hydroxide and 50 ml of water).
Immunofluorescence was performed using the following primary antibodies: rat anti-CD45 for total leukocytes (1:100; ThermoFisher Sci., Rockford, IL, USA cat. #MA1-81247), rat anti-MBP for myelin basic protein (ThermoFisher Sci.; 1:100; cat. #MA1-24990), rabbit β-APP for β-amyloid peptide (Thermo Fisher Sci.; 1:200 cat. # 36-6900).
In short, the sections were preincubated for 40 min in PBS containing 5% normal goat serum (Sigma-Aldrich, St. Louis, MO, USA), 0.01% sodium azide, 0.05% thimerosal, 0.1% bovine serum albumin, 0.5% Triton X-100, and 2% dry milk. They were next incubated overnight at room temperature with primary antibodies and after a rinse in PBS incubated for 90 min with the secondary Cy3-conjugated goat anti-rat antiserum (Jackson IR, West Grove, PA, USA; 1:300, cat. #112-165-167), goat anti-rat Alexa488-conjugated antibodies (Jackson IR, West Grove, PA, USA; 1:100; cat. #112-545-167) or goat anti-rabbit Alexa488-conjugated antibodies (Jackson IR, West Grove, PA, USA; 1:100, cat. #111-545-144). Then, sections were washed three times in PBS and mounted in glycerol/PBS solution. DAPI staining (ThermoFisher Sci., Rockford, IL, USA; 1.5 ug/ml; cat. #62248) was used to visualize cell nuclei.
Microscopy, morphometry and image collection
Spinal cord sections were examined using Olympus BX50 brightfield/epifluorescence microscope (Olympus, Tokyo, Japan). All images were recorded using DP 71 Olympus camera (1360×1024 pixels), stored as TIFF files and processed for quantitative analysis using ImageJ software (NIH, Bestheda, Maryland, USA).
To quantify the degree of neuropathological changes in the spinal cord, all the recorded images of spinal cords were analyzed. EAE severity was assessed by examination of focal lesions that show inflammatory cell infiltration (mainly leukocytes and macrophages) in spinal cord sections. The number of inflammatory lesions/aggregates of spinal cord was counted per cross section. Twelve sections per animal were examined (60 images per experimental group and disease phase, 5 mice per group/phase).
Inflammation, demyelination, axonal damage were quantified as the percentage of infiltrated/demyelinated/axonal damage areas against the total area of the whole spinal cord section. HE staining demonstrated cellular infiltrations. The recorded images were used to assess the degree of inflammation by calculating the percentage of infiltration area of the whole spinal cord cross-sectional area occupied by the gray and white matter. In contrast, demyelination and axonal damage were seen as brighter areas in the white matter of the spinal cord. The percentage of their surface area was related to the degree of demyelination/axonal damage in the entire spinal cord. Twelve sections per animal were examined (60 images per experimental group and disease phase, 5 mice per group/phase).
The percentage of immunopositive areas present in the whole spinal cord cross-sections (white and gray matter) were assessed in images for each antigen in the successive phases of EAE. The expression of CD45, MBP and beta-amyloid precursor protein (β-APP) was quantified in micrographs taken at the same magnification by measuring the area of immunopositive structures in a single image and expressed as a percentage of the total area of the spinal cord. A total of 30 images were assessed per each experimental condition (experimental group and disease phase, 5 mice per group/phase, 6 sections per mouse).
Data analysis
GraphPad Prism 5.0 software (GraphPad, La Jolla, CA, USA) was used throughout this study for statistical analyses. All values were expressed as mean ±standard error of the mean (SEM). Statistical significance was verified using ANOVA with Bonferroni multiple comparison test. P<0.05 was regarded as statistically significant.
RESULTS
Clinical experimental autoimmune encephalomyelitis progression
The outline of the study is presented in Fig. 1b. In MOG35–55 immunized mice, the first symptoms of EAE appeared on post-immunization day 9. On the basis of the applied scoring protocol, three distinct phases of the disease were distinguished: onset, peak and chronic. In the onset phase, the clinical score increased to 1.1±0.1, reached the highest value 3.1±0.1 in the peak phase and then decreased to 1.7±0.2 in the chronic phase (Figs. 1a and 2a). All differences between the phases were significant. Control mice did not develop any symptoms of the disease (Fig. 1a).

Inflammatory infiltrations assessed by hematoxylin/eosin staining and CD45 expression
HE staining allowed to assess the number of inflammatory lesions and area of inflammatory infiltrations in the spinal cord per group and phase (Figs. 2b and 3j). The lesions were located in the white matter. DAPI staining demonstrated higher density of spinal cord cells compared to control samples (Fig. 4a-4c).
The number of inflammatory lesions increased from 2.37±0.11 in the onset phase to 9.08±0.25 in the peak phase and then slightly decreased to 7.08±0.20 in the chronic phase (Fig. 2b). In EAE mice, the temporal pattern of inflammation changes was similar to that of EAE progression expressed by clinical scores (Figs. 1a, 2a, 2b and 3j). The inflammatory foci were located in similar, repetitive areas of the spinal cord (Fig. 1c) and often colocalized with areas of demyelination and axonal damage.
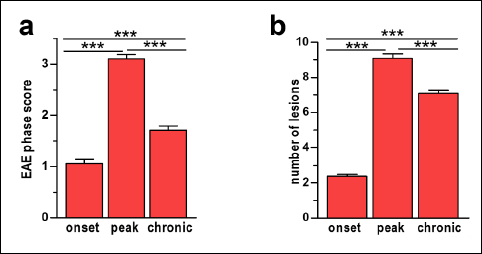 |
Fig. 2. Diagrams show the mean clinical scores in experimental autoimmune encephalomyelitis (EAE) phases (a) and number of inflammatory lesions (b). In (a) and (b) data are presented as means ±SEM; n=5 per group. Statistical significance was verified using ANOVA with Bonferroni multiple comparison test at 0.05 confidence level (***p<0.001; **p<0.01; *p<0.05). |
The infiltration of leukocytes into the spinal cord was observed in many areas of the white matter (Fig. 1c and 3d). Inflammation area gradually increased from 2.24±0.14% in the onset phase to reach the maximum 11.07±0.45% in the peak phase of EAE and inflammation slightly declined to 5.29±0.27% in the chronic phase (Fig. 3j). In all phases of the disease, leukocytes formed local aggregates with high cell density.

To further confirm inflammatory cell infiltration, spinal cord sections were assessed by measuring the expression of CD45 antigen, a general marker of proinflammatory leukocytes. Immunostaining for CD45 confirmed the location of inflammatory foci predominantly in white matter, as demonstrated by HE staining, but also showed individual leukocytes in white matter outside of inflammatory infiltration and in gray matter (Fig. 4d). The values and temporal pattern of CD45 expression: increase from the onset phase (1.74±0.17%) to peak phase (10.15±0.50%), and then decrease in the chronic phase (6.23±0.45%) were similar to those obtained for HE staining (Fig. 4g).
Control mice demonstrated normal histology without the presence of inflammatory cell infiltrates in both HE stained (Fig. 3a) and CD45 immunofluorescence labeled samples (not shown).
Assessment of demyelination by Luxol Fast Blue staining and myelin basic protein expression
Histopathological examination of spinal cord tissue also included LFB staining to visualize and quantify the degree of demyelination (Fig. 3b, 3e, 3h and 3k), characteristic for both EAE and MS. The level of demyelination progressed from the onset phase (0.54±0.14%) to the peak phase (5.15±0.31%) (Fig. 3k) and was associated with the peak of EAE symptoms and inflammation. However, as EAE progressed, demyelination remained at the same level between peak and chronic phases (Fig. 3k). Spinal cords of control mice did not contain areas of demyelination (Fig. 3b).
Along with the assessment of demyelination, expression of myelin basic protein (MBP), a structural protein of myelin (Fig. 4e) confirmed association of myelin damage with the progression of EAE. Immunofluorescence labeling of MBP displayed its gradual decrease with progression of EAE severity: from 22.19±0.49% in the onset phase to 15.06±0.30% in the peak phase and to 10.83±0.34% in the chronic phase (Fig. 4h). Control mice showed MBP expression similar to that recorded in the onset phase of the EAE mice (not shown).
 |
Fig. 4. Representative immunofluorescence images of DAPI (a-control, b-experimental autoimmune encephalomyelitis (EAE) mice, c-high magnification of area enclosed within yellow square) and images of CD45, myelin basic protein (MBP) and beta-amyloid peptide (β-APP) expression in the peak phase of EAE severity (d-f). CD45, MBP and β-APP content in the spinal cords of EAE mice (g-i). In micrographs (d-f), yellow dashed line marks the border between white and gray matter. Expression of the studied antigens was quantified as the percentage of the immunostained area (g-i). Data are presented as means ±SEM; n=5 per group. Statistical significance was verified using ANOVA with Bonferroni multiple comparison test at 0.05 confidence level (***p<0.001; **p<0.01; *p<0.05; ns, not significant). Magnification is indicated by scale bars (a, b = 500 µm, c-f = 100 µm). |
Evaluation of axonal damage by Bielschowsky silver impregnation staining and beta-amyloid precursor protein expression
The third main characteristics of EAE and MS is axonal damage, visualized by BSI staining (Fig. 3c, 3f, 3i). A gradual increase in axonal damage was evident throughout the course of EAE, from 1.41±0.10% in the onset phase to 5.95±0.32% in the peak phase and to 10.62±0.23% in the chronic phase (Fig. 3l).
β-APP, the major component of amyloid plaques in neurodegenerative diseases, promotes axonal damage. β-APP accumulation in the white matter of the spinal cord (Fig. 4f) was observed already in onset phase of EAE (0.67±0.06%), increased in the peak phase (1.89±0.20%) and remained at a similar level in the chronic phase (2.02±0.16%), (Fig. 4i). Samples collected from control mice showed neither axonal damage (Fig. 3c) nor the presence of β-APP (not shown).
DISCUSSION
The inflammation and neurodegenerative processes that contribute to the pathology of MS/EAE are a complex interplay between autoreactive lymphocytes migrating across the brain-blood barrier and CNS resident cells, it subsequently leads to axonal and neuronal damage. The pathogenetic parameters of EAE/MS: inflammation, demyelination and axonal damage can be identified by histopathological and immunohistochemical stainings (2, 3, 7) and quantified by image analysis techniques. In this study, we assessed these parameters during progression of EAE, which according to clinical scores can be divided into three successive phases: onset, peak and chronic. The investigated parameters were correlated with EAE severity, characterized by neurological symptoms.
CD45, a general marker of proinflammatory leukocytes, showed a temporal pattern similar to that of the clinical scores. As demonstrated by HE staining, the inflammatory foci were predominantly located in white matter, but individual leukocytes were also observed in gray matter. During the chronic phase of EAE, inflammatory changes in the gray matter can lead to muscle weakness and paralysis (12). T cells and their main subsets, Th1 and Th17 present in the inflammatory infiltrations play an important role in MS and EAE progression. They were observed in demyelinating lesions and became activated in the relapsing stage of MS (13, 14). Our earlier study demonstrated in the inflammatory foci a high density of T and B lymphocytes, neutrophils and monocytes/macrophages contributing to the pathogenesis of MS/EAE (8).
Demyelination demonstrated by LFB staining increased in the peak phase of EAE and remained at the similar level in the chronic phase. Recruited monocyte-derived macrophages (together with CNS-resident microglia) are known to phagocytize the myelin sheaths resulting in demyelination (15). Also, active CD4 T cells may induce demyelination (16). On the other hand, stabilization of demyelination process seems to be associated with decrease in the inflammation intensity in the chronic phase of EAE. LFB staining in the chronic phase of EAE could also suggest partial remyelination of the damaged axons, however, other studies demonstrated that the remyelination process occurred at a very late stage of EAE progression, after 6 months (17, 18) and in the present study the assessment of spinal cord samples in the chronic phase of EAE was done on day 30, before the onset of remyelination. Immunostaining showed a gradual decrease in expression of MBP, a major protein constituent of the myelin sheaths, in the successive phases of EAE. This result is partly discrepant with LFB staining which did not show further increase in demyelination in the chronic phase of EAE. LFB staining mechanism is based on an acid-base reaction with myelin lipoproteins (19), hence it seems to be less precise than MBP immunostaining.
Axonal damage is a key component of EAE progression and leads to permanent neurological disability (3, 8, 17). With persistent inflammation as well as impaired remyelination, axons degenerate, contributing to CNS atrophy and exacerbation of the disease (19-21). BSI staining showed that the level of axonal damage gradually increased from the onset phase, through peak, and reached the maximum in the chronic phase. These results do not agree with the data presented by Recks et al. (20) who found that axonal damage coincided with the initial clinical symptoms and remained stable in the later phases of the disease. However, the authors of that study applied a different timeline of the experiment, not corresponding with the presently accepted phases of EAE: they examined mice on the first day of EAE symptoms, and then three and six months later, hence their results cannot be compared with ours.
β-APP is regarded as a marker of axonal damage (22-25). Its expression demonstrated progress in the onset and peak phases of EAE and stabilization in the chronic phase. Again, this result did not fully correspond with those obtained in BSI stained samples: β-APP expression did not increase in the chronic phase of EAE. Increased expression of β-APP can enhance remyelination in MS (24). In EAE, the treatment with b-amyloid decreased the proliferation of certain types of T lymphocytes and reduced the production of pro-inflammatory cytokines, such as interferon-γ (25).
Partly discrepant results of classical stainings (LFB, BSI) and immunohistochemical detection of markers in case of myelin sheaths (MBP) and injured axons (β-APP) provoke the question whether both approaches can be regarded as equally adequate in the examination of central nerve tissue samples in EAE and other neurodegenerative diseases. Further studies should provide the answer.
Histopathological stainings have been used in many studies to examine anti-inflammatory/neuroprotective effects of drug treatment in EAE/MS (9, 10, 26). Several systemic immunosuppressive or immunomodulatory drugs such as interferon-beta (IFN-β), glatiramer acetate, mitoxantrone, fingolimod, and natalizumab were used to treat or delay EAE/MS (9, 10, 27-30). For example, natalizumab inhibits activated Th1 cell trafficking across the BBB (9, 10, 28, 29). Glatiramer acetate reduces T cell activation and differentiation to Th1 subset as well as T cell reactivation and induction of T cell-mediated demyelination (28, 29).
Our previous studies also showed that in EAE, the histopathological changes observed in the spinal cord were accompanied by changes in white matter stiffness, which could be considered as an early signature of EAE (11). They also demonstrated the effect of anti-VLA-4 mAb treatment (natalizumab) on the number and density of inflammatory infiltrations, expression of adhesion molecules, as well as on the level of metalloproteinases and their tissue inhibitors in the course of the disease (9, 10).
Classical histological stainings combined with immunohistochemistry allow for the assessment of the condition of the nervous tissue and its possible abnormalities (6-8, 31). Our studies showed that all the analyzed histopathological parameters change in the initial phase of EAE and increase in parallel with the severity of EAE. T-cell influx generates inflammatory lesions which induce demyelination and axonal injury in the spinal cord. Although inflammation intensity subsides in the chronic phase of EAE, the neurodestructive processes induced by inflammation: demyelination and axonal damage continue in that phase.
The temporal patterns of inflammatory infiltration into the spinal cord and of the resulting neurodegeneration in the course of MS and EAE can provide valuable insights into the pathogenesis of these diseases and stimulate therapeutic strategies in MS aimed at prevention or inhibition of penetration of inflammatory cells across the blood-brain barrier.
Authors contribution: GPF designed the experiments, carried out the immunization of mice and histological examinations; GPF, JAL wrote the manuscript; MF, GPF, JAL were involved in data analysis; GPF, BW provided histological examination. All authors contributed to interpretation of the results and to discussion.
Acknowledgments: The study was supported by the grant N41/DBS/000950 from the Jagiellonian University Medical College to GPF.
Conflicts of interest: None declared.
REFERENCES
- Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 2001; 164: 1079-1106.
- Voskuhl RR, MacKenzie-Graham A. Chronic experimental autoimmune encephalomyelitis is an excellent model to study neuroaxonal degeneration in multiple sclerosis. Front Mol Neurosci 2022; 15: 1024058. doi: 10.3389/fnmol.2022.1024058
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000; 123: 1174-1183.
- Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol 2017; 133: 223-244.
- Gold R. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006; 129: 1953-1971.
- Steinbach K, Merkler D. Neuropathological techniques to investigate CNS pathology in experimental autoimmune encephalomyelitis (EAE). Methods Mol Biol 2016; 1304: 189-209.
- Gibson-Corley KN, Boyden AW, Leidinger MR, et al. A method for histopathological study of the multifocal nature of spinal cord lesions in murine experimental autoimmune encephalomyelitis. PeerJ 2016; 4: e1600. doi: 10.7717/peerj.1600
- Pyka-Fosciak G, Stasiolek M, Litwin JA. Immunohistochemical analysis of spinal cord components in mouse model of experimental autoimmune encephalomyelitis. Folia Histochem Cytobiol 2018; 56: 151-158.
- Pyka-Fosciak G, Lis GJ, Litwin JA. Effect of natalizumab treatment on metalloproteinases and their inhibitors in a mouse model of multiple sclerosis. J Physiol Pharmacol 2020; 71: 265-273.
- Pyka-Fosciak G, Lis GJ, Litwin JA. Adhesion molecule profile and the effect of anti-VLA-4 mAb treatment in experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Int J Mol Sci 2022; 23: 4637. doi: 10.3390/ijms23094637
- Pyka-Fosciak G, Zemla J, Lis GJ, Litwin JA, Lekka M. Changes in spinal cord stiffness in the course of experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Arch Biochem Biophys 2020; 15: 108221. doi: 10.1016/j.abb.2019.108221
- Milovanovic J, Arsenijevic A, Stojanovic B, et al. Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol 2020; 11: 947. doi: 10.3389/fimmu.2020.00947
- Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018; 97: 742-768.
- Steinman L. The discovery of natalizumab, a potent therapeutic for multiple sclerosis. J Cell Biol 2012; 199: 413-416.
- Weinstock-Guttman B, Nair KV, Glajch JL, Ganguly TC, Kantor D. Two decades of glatiramer acetate: from initial discovery to the current development of generics. J Neurol Sci 2017; 376: 255-259.
- Hendrickx DA, Schuurman KG, van Draanen M, Hamann J, Huitinga I. Enhanced uptake of multiple sclerosis-derived myelin by THP-1 macrophages and primary human microglia. J Neuroinflammation 2014; 11: 64. doi: 10.1186/1742-2094-11-64
- Wu J, Ohlsson M, Warner EA, et al. Glial reactions and degeneration of myelinated processes in spinal cord gray matter in chronic experimental autoimmune encephalomyelitis. Neuroscience 2008; 156: 586-596.
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 2010; 162: 1-11.
- Pearse AGE. Histochemistry, Theoretical and Applied. London, Churchill Livingstone 1972.
- Recks MS, Stormanns ER, Bader J, Arnhold S, Addicks K, Kuerten S. Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin Immunol 2013; 149: 32-45.
- Klotz L, Antel J, Kuhlmann T. Inflammation in multiple sclerosis: consequences for remyelination and disease progression. Nat Rev Neurol 2023; 19: 305-320.
- Ghosh N, DeLuca GC, Esiri MM. Evidence of axonal damage in human acute demyelinating diseases. J Neurol Sci 2004; 222: 29-34.
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120: 393-399.
- Matias-Guiu JA, Oreja-Guevara C, Cabrera-Martin MN, Moreno-Ramos T, Carreras JL, Matias-Guiu J. Amyloid proteins and their role in multiple sclerosis. Considerations in the use of amyloid-PET imaging. Front Neurol 2016; 7: 53. doi: 10.3389/fneur.2016.00053
- Jones R. Neurological disorders: Two sides to b-amyloid. Nat Rev Neurosci 2012; 13: 666. doi: 10.1038/nrn3336
- Ge ZZ, Wu YB, Xue ZY, Zhang K, Zhang RX. The therapeutic effects of the peptidyl-prolyl cis/trans isomerase Pin1 inhibitor juglone on animal-model experimental autoimmune encephalomyelitis. J Physiol Pharmacol 2021; 72: 195-202.
- Farooqi N, Gran B, Constantinescu CS. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J Neurochem 2010; 115: 829-844.
- Hart FM, Bainbridge J. Current and emerging treatment of multiple sclerosis. Am J Manag Care 2016; 22: 159-170.
- Amin M, Hersh CM. Updates and advances in multiple sclerosis neurotherapeutics. Neurodegener Dis Manag 2023; 13: 47-70.
- Adamczyk-Sowa M, Sowa P, Adamczyk J, et al. Effect of melatonin supplementation on plasma lipid hydroperoxides, homocysteine concentration and chronic fatigue syndrome in multiple sclerosis patients treated with interferons-beta and mitoxantrone. J Physiol Pharmacol 2016; 67: 235-242.
- Wuerch E, Mishra M, Melo H, Ebacher V, Yong VW. Quantitative analysis of spinal cord neuropathology in experimental autoimmune encephalomyelitis. J Neuroimmunol 2022; 362: 577777. doi: 10.1016/j.jneuroim.2021.577777
A c c e p t e d : August 31, 2023