THE CYCLOOXYGENASE-2/PROSTAGLANDIN E2 PATHWAY AND ITS ROLE IN THE PATHOGENESIS OF HUMAN AND DOG HEMATOLOGICAL MALIGNANCIES
INTRODUCTION
Cyclooxygenases (COXs, also known as prostaglandin endoperoxide synthases or prostaglandin G/H synthases) and PGs have emerged in recent years as important players in cancer progression (1, 2). Overexpression of COX-2 leads to an increase of PGE2 in cancer microenvironment. It is well documented that in the progression of colorectal cancer, dysregulation of COX-2/PGE2 pathway participates in tumour initiation, promotes tumour maintenance and the formation of metastases (3, 4). This pathway suppresses the innate and adaptive immune responses leading to development of solid tumours (5, 6). Overexpression of COX-2 promotes polarization of M1 macrophages to a pro-tumour M2 type (7, 8). PGE2 receptors displayed on NK cells contribute to the inhibition of their migration and production of interferon-γ (9, 10). The COX-2 overexpression in cancer microenvironment inhibits maturation of dendritic cells and reduces the expression of MHC class II molecule (11, 12).
In various cancers, an increased activity of T regulatory lymphocytes (Tregs) represents one of the strategies by which a tumour escapes immune surveillance. The influence of PGE2 on Tregs has been demonstrated in a mouse model of lung cancer in which COX-2 overexpression and subsequent production of significant amounts of PGE2 augmented Treg numbers (13-15). It has been found that increased expression of COX-2 in neoplastic cells enhances the expression of FoxP3 (forkhead box P3 transcription factor) in Tregs, which potentiate their suppressive activity in vitro (16, 17). It has also been documented that PGE2 inhibits proliferation of B and T lymphocytes, as well as apoptosis and cytokine production by CD4+ T cells (1).
However, many questions concerning the mechanisms of COX-2/PGE2 influence on suppression of innate and adaptive immune system in hematological malignancies remain unanswered.
PROSTAGLANDINS AND CYCLOOXYGENASES
Prostaglandins (PGs) are small lipid-derived molecules that regulate numerous physiological processes: ovulation, release of neurotransmitters, platelets aggregation and contribute to cytoprotection of gastric mucosa (1, 18, 19). They also play a role in many pathological processes, including cancer progression. COXs are enzymatic inflammatory regulators responsible for production of all prostaglandins. They convert arachidonic acid primarily to prostaglandin G2 (PGG2), which is then followed by the reduction of PGG2 to prostaglandin H2 (PGH2). The latter form is subsequently converted by cell-specific prostaglandin synthases (PGEs) into various, biologically active PGs: PGI2, PGD2, PGE2 and PGF2α (1, 13, 20, 21).
The action of prostaglandins is mediated by binding to the cell specific membrane surface G-protein-coupled receptors: EP1 - EP4 (11, 18). There are species differences in prostaglandin receptors (EP). In humans, there are seven splice variants of EP3, while in mice the same receptor consists of three different isoforms: α, β and γ (1). The specific role of each EP receptor in cancer is still under investigation. It has been shown, that EP receptors are engaged in intestinal tumorigenesis - EP2 is involved in intestinal polyps growth promotion and contribute to enhanced angiogenesis (20). The density of EP4 receptor on colonic epithelial cells is 100-fold higher in colon cancer compared to normal colonic epithelium (11). Cell activation mediated by EP4 receptor occurs primarily through the PI3K (phosphatidylinositide 3-kinase) dependent pathway and leads to the phosphorylation of extracellular signal-regulated kinases (ERKs), followed by the induction of early growth response factor 1 (EGR-1) (22, 23). Cyclin D1, which is required for passing through G1 phase of the cell cycle is controlled by EGR-1. Thus, EP4 activation by PGE2 stimulates proliferation of colorectal carcinoma cells (24, 25). In mice, EP2 receptors are involved in skin tumour development (18). Homozygous deletion of the gene encoding EP2 receptor in a mouse model of human familiar adenomatous polyposis (ApcΔ716 mice) reduced the number and size of intestinal polyps because of decreased PGE2 production (26, 27). Functional EP3 and EP4 receptors are also expressed on the nuclear envelope, for instance of the endothelial cells, and modulate the transcription of a gene which encodes inducible nitric oxide synthase, suggesting that they are implicated in the regulation at the nuclear level (1). Nevertheless, further studies are required to determine their role and functions in this location.
Three distinct isoforms of COX have been recognized. The first isoform - COX-1 is an enzyme constitutively expressed in a broad range of cells and tissues e.g. renal cortex, stomach mucosa and also on platelets and vascular endothelium. It is responsible for the synthesis of eicosanoids that maintain essential functions of the digestive, urinary and cardiovascular system. The second isoform - COX-2, is an inducible enzyme and typically is not expressed in resting cells, but it can be induced in response to cytokines associated with inflammatory diseases and carcinogenesis (13). The expression of COX-2 is increased in many types of cancers (mammary, prostate, colon, hematological malignancies) compared to normal cells (11, 13). This isoform presumably participates in the promotion of angiogenesis, tissue invasion of tumour cells and tumour resistance to apoptosis (28, 29). The third - COX-3 isoform was identified in humans, mice and dogs and it is the most abundant in cerebral cortex (18, 30-32). Its expression in primary brain tumours has been examined in mice glioblastoma models. Both in vitro and in vivo experiments in that models proved a correlation between COX-3 expression and glioblastoma development (30).
The increase in COX expression and subsequent increase in PG production in neoplasia suggest that they play an important role in cancer development. Thus their influence on carcinogenesis is investigated in vivo and in vitro in many solid tumours (5, 12, 17, 18, 27). However, the role of COX and PGs in hematological malignancies remains unexplained and needs further investigation.
CYCLOOXYGENASE-2/PROSTAGLANDIN E2 ACTIVATION IN CANCER
The role of COX-2/PGE2 on immune evasion has been frequently investigated in many types of cancer. Tumour microenvironment consist of extracellular matrix, blood and lymphatic vessels, fibroblasts, as well as many different types of the immune cells (e.g. lymphocytes, macrophages, dendritic cells). In many cancer, tumour cells secrete PGE2 to tumour microenvironment to promote tumour growth but also inhibit anti-cancer immunity (33). Depending on T lymphocytes maturation stage, COX-2/PGE2 pathway influences their proliferation, apoptosis and release of cytokines. PGE2 inhibits proliferation of CD4+CD8+ thymocytes and immature B cells (1). Increased concentration of PGE2 in tumour microenvironment also inhibits the proliferation of CD4+ and CD8+ T cells and impairs their functions (Fig. 1). Overexpression of COX-2 and increased PGE2 levels in the tumour microenvironment have an impact on polarization of macrophages towards tumour associated macrophages (TAMs) phenotype, and the increased infiltration of macrophages and Tregs in the tumour tissue (11). Myeloid derived suppressor cells (MDSCs) represent heterogeneous population of myeloid cells at different stages of differentiation. They are significant cellular players in cancer environment able to suppress the functions of T and NK cells (34). Many different subsets of MDSCs which are increased in cancer have been identified both in the tumour tissue and in blood of cancer patients (35, 36). The phenotype of dominant MDSC subpopulation presumably depends on the type of tumour. MDSCs are arrested in the immature phase, under the influence of specific signalling network which includes inflammatory mediators such as IL-10 and PGE2. It has been shown that elevated concentration of COX-2 and close contact with cancer cells can contribute to the increase in the number of MDSC in the tumour tissue (11, 37, 38).
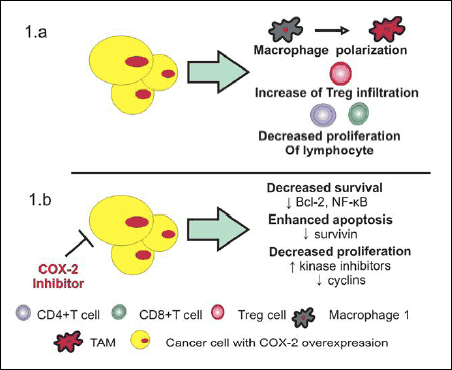 |
Fig. 1. Influence of COX-2/PGE2 pathway (1a) and COX-2 inhibitors (1b) in tumour microenvironment. (1a) Cancer cells with COX-2 overexpression induce polarization of M1 macrophages to tumour associated macrophages (TAMs) that infiltrate tumour tissue. COX-2 overexpression and increase concentration of PGE2 inhibit proliferation of CD4+ and CD8+ cells as well as enchance Treg infiltration into tumor microenvironment. (1b) COX-2 inhibitors decrease cancer cell survival by an inhibition of NF-κB and antiapoptotic protein Bcl-2. They are also responsible for reduction in survivin concentration, thus enhance apoptosis of neoplastic cells. Additionally, COXIBs inhibit cancer cell proliferation as a result of cyclins downregulation and increased activity of kinase inhibitors. |
Tumorigenesis partially results from an imbalance between cell proliferation and apoptosis. In many type of cancer the COX-2/PGE2 pathway regulates both cell proliferation and apoptosis. Data obtained from studies on acute promonocytic leukemia and human myeloblastic leukemia cell lines have shown that a specific COX-2 inhibitor reduces proliferation of cells and arrest them in G0/G1 phase (39, 40). A similar effect was observed after in vitro administration of celecoxib into three cell lines of human Burkitt’s lymphoma (41).
In chronic myeloid leukemia celecoxib reduced proliferation of cells, that was correlated with the downregulation of cyclin D1 and E and upregulation of kinase inhibitors: p16 and p27, which prohibit cells progression from G1 phase to S phase (41).
NF-κB is an important regulator of apoptosis. It directly activates antiapoptotic proteins: c- IAPP1 (e.g. survivin), c-IAP2 and ICAP associated with TNF receptor but also Bcl-2 protein homologues. It has been shown that NF-κB is constitutively activated in various neoplastic cell types allowing them to evade apoptosis (42, 43). The activation of NF-κB requires the serine - threonine IκB kinase (IKK). Therefore, as a therapeutic target, it is necessary to block activity of IKK in order to inhibit of NF-κB (44, 45). Inhibition of IKK renders cancer cells more sensitive to apoptosis (46). Among a wide group of non-steroidal anti-inflammatory drugs, some (acetylsalicylic acid, ibuprofen, or sulindac) have shown an inhibitory effect on IKK (47, 48). A study made by Chen et al. showed that COX-2 overexpression in human lung cancer cells stabilized apoptosis inhibitor- survivin. Celecoxib inhibited IKK activity which resulted in decreased survivin expression and increased cell apoptosis (49).
Therapeutic resistance (TR) in many cancers is one of the major challenges faced by patients and oncologists. One of the most discussed nowadays theory of TR is involvement of cancer stem cells (CSCs) in that process, which has been identified in many neoplasia. Leukemic CSCs- are critical in the initiation and maintenance of leukemia, but presence of similar cell populations that may generate B or T-cell lymphoma upon mutation remains uncertain (33, 50). CSCs well tolerate hypoxia, inflammation in tumour microenvironment and evade cytotoxic effect of chemotherapy and become the important cause of the TR. COX-2 overexpression observed in CSCs, and regulation of COX-2/PGE2 axis by miRNAs (upstream-related miRNA-27a and downstream-related miRNA -30a,-101b) shows that inhibition of COX-2/PGE2 axis could be potential therapeutic target in cancer therapy in controlling repopulation of CSCs (33, 51).
Other cells involved in tumorigenesis are platelets (PLTs). They role in maintaining of tumour growth and metastasis, via cancer cell- platelet interactions is well documented. Tumour cells induce secretion thrombin, cathepsin B, MMP-2 and 14 from platelets α-granules (52, 53). During PLTs aggregation while cancer cells contact, numerous of angiogenesis regulators (vascular endothelial growth factor, basic fibroblast growth factor) are released promoting tumour angiogenesis (52, 54). Also during activation and aggregation PLTs shed submicron structures- platelets microparticles (PMPs). Based on their size, they are named exosomes (40 – 100 nm) or ectosomes (0.1 µm) (52, 54, 55). Many in vivo and in vitro experiments show that PMPs after fusion with donor cells changes their properties using three mechanisms. First way is to donate maternal adhesive glycoproteins and membrane receptors (CD41/61, CD62P) into donor cell, second is stimulation by ligands to produce cytokines and growth factors (55, 56). The third mechanism is reprograming transcription in donor cells via miRNAs and other transcriptomic factors from PMPs (57). In many cancers (prostate, colon, breast, lung cancers) PMPs number increase and it is a poor prognostic factors. Hu et al. reported expression of COX-1 and COX-2 on PMPs and platelets (58). Considering the role of PMPs in cancer invasiveness and metastasis it is now discussed how valid is the COXIBs anticancer therapy in decreasing upregulation of COX-2 in cancer cells after fusion with PMPs.
SUPPRESSION OF CYCLOOXYGENASE-2/ PROSTAGLANDIN E2 AXIS AS A PART OF ANTICANCER STRATEGY IN HEMATOLOGICAL MALIGNANCIES
The role of COX-2/PGE2 axis in tumour development and cancer therapeutic resistance has been proved in numerous studies during few decades. Most of works estimate this axis in solid tumours, but also in hematological malignancies it plays a crucial role. In this section we discuss the suppression of COX-2/PGE2 axis in hematological malignancies as a potential strategy in anticancer therapy. This can be achieved by increasing cancer cells apoptosis dependently or independently of the PGE2/COX-2 or by downregulation of EP receptors. Table 1 shows COX-2 inhibitors and EP receptor antagonists and their modulatory influence on cell lines and hematological malignant cells from human patients.
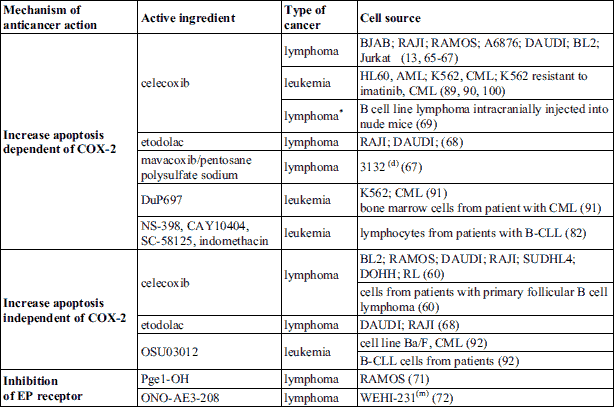
Lymphomas
Non-Hodgkin’s lymphomas (NHLs) represent a heterogeneous group of hematological malignancies derived from B, T lymphocytes or NK cells. Diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) are the most frequent lymphoma types in human and represent the sixth most common cause of cancer-related deaths in the occidental countries (59-62). In Burkitt’s lymphoma (BL) cell lines, compared to normal blood B cells, COX-2 may be phosphorylated and its expression is always elevated (63, 64). In vitro studies performed on lymphomas cell lines confirmed influence of COX-2 on apoptosis intensification. Jendrossek et al. have used two lymphoma cell lines with and without death receptor CD95 and with a defect in mitochondrial signalling pathways to evaluate influence of celecoxib treatment on apoptosis. It was concluded that celecoxib induces apoptosis via novel apoptosome-dependent, but Bcl-2-independent mitochondrial pathway (65, 66). Runder et al. have suggested that pro-apoptotic Bak protein is sequestered by Bcl-xL and Mcl-1 is downregulated in Jurkat T lymphoma cells as an effect of celecoxib treatment (67). The differences in binding of Bcl-2 and Bcl-xL with other Bcl-2 family members observed in the experiment have indicated that Bcl-xL and Bcl-2 use different mechanisms to protect from apoptosis in response to distinct stimuli (67). In BL cell lines, celecoxib reduces cell proliferation negatively influencing expression of cyclin A and B and induces apoptosis in more than 85% cells (13). The study on the effect of etodolac on BL cell lines lacking COX-2 expression has shown that induction of apoptosis resulted from a decrease of Bcl-2 levels (68). It was also confirmed using a mouse model of central nervous system lymphoma in vivo (13, 69).
Increased cancer cells apoptosis under influenced of COXIBs could be independent of their PGE2/COX-2 axis inhibition. It was demonstrated that treatment with celecoxib reduces PGE2 production by stromal cells in follicular lymphoma (60). Co-culture of stromal cells with primary follicular lymphoma B cells and NHL B-cell lines confirms that celecoxib increases the apoptotic activity independently of the PGE2/COX-2 axis but via increased activity of TNF-related apoptosis-inducing ligand (TRAIL) (60, 70).
EP4 receptor in immature/mature B cells inhibits their survival via NF-κB. Gobec et al. evaluate the effectiveness of Pge1-OH, a specific EP4 receptor agonist in BL cell line (Ramos cells). This causes increased cells sensitivity to doxorubicin and bortezomid as effect of decreased NF-κB activity in Ramos cells (71). In immature B cells PGE2 and EP4 receptor plays important role as modulator of B cells response. ONO-AE3-208 EP4 receptor antagonist on immature B cell lymphoma (WEHI 231 cells) stops cells in G0/G1 and induce caspase mediated apoptosis (72).
Lymphomas account for about 20% of all canine neoplasms and about 85% of lymphoid malignancies, of which the most common is DLBCL. There are only a few papers evaluating PGE2 concentration or COX-2 expression and their role in canine lymphoma (73). Rodrigeus et al. have examined lymph nodes obtained from dogs with multimeric lymphomas and healthy controls. COX-2 immunoreactivity was not detected in all normal lymph nodes but only in a few samples of canine lymphoma (74). Asproni et al. have examined a larger group of normal, hyperplastic and neoplastic canine lymphoid tissues and elevated levels of COX-2 were found in 30% of lymphoma tissues, with the same values in T and B cell lymphomas (75). In another study, an impact of carprofen and mavacoxib on the survival of canine cancer cells and cancer stem cells (CSCs) (3 osteosarcoma and lymphoma, mast cell tumour and haemangiosarcoma cell lines) has been compared (76, 77). In all tested cell lines there was an increase of Bax protein levels and mavacoxib increased apoptosis in caspase-2, 3, 8 and 9 independent way. Mavacoxib has a cytotoxic effect on CSCs from osteosarcoma cell lines and retards cell migration in all cancer cell lines (76).
Leukemias
Chronic lymphocytic leukemia (CLL) is an uncontrolled growth of mature B lymphocytes that are highly proliferative and resistant to many chemotherapeutic drugs and it constitutes approximately one third of all leukemias (78-81). Compared to normal B lymphocytes, expression of COX-2 was higher in analysed B-CLL samples (13).
It was reported that in contrast to naïve B cells, B-CLL cells constitutively express COX-2 and produce PGs which are important for their survival (82, 83). Secchiero et al. demonstrated, that cells isolated from B-CLL patients treated by COXIB (NS-398) are sensitive to TRAIL dependent apoptosis. Additionally, the cells sensitivity to a cytotoxic effect of chlorambucil, increases in about 50% after pre-treatment with (NS-398) (84).
There are few articles ascribing effect of COXIBs analogues which cannot inhibit COXs in hematological malignancies. Johnson et al. examined the effect of a low concentration of the third generation derivative of celecoxib (OSU03012) on B-CLL cells in vitro and showed that it mediates apoptosis in B-CLL cells by a caspase independent mechanism (85).
Chronic myeloid leukemia (CML) is a clonal bone marrow stem cells disorder in which the proliferation of mature granulocytes and their precursors was found and it is an effect of chromosomal translocation and fusion of the BCR and ABL genes, resulting in an unrestrained proliferation (13, 86-88). CML cells expressed increased levels of COX-2 compared to healthy cells and it correlated with a poor prognosis (13, 89).
Just as observed in lymphomas in vitro works on CML cell lines increase cancer cells apoptosis dependently or independently of the PGE2/COX-2. Subhashini et al. examined the influence of celecoxib on chronic myelogenous leukemia cell line (K562) and its proapoptotic effect was a result of the release of cytochrome c into the cytoplasm, inhibition of NF-κB and decreased level of Bcl-2 without changes in Bax concentration (90). Another COXIB (DuP-697) also inhibited proliferation of K562 cells and bone marrow CML cells from CML patients, and induced apoptosis via caspase 8 activation (91). Tseng et al. examined OSU-03012 effect in vitro on CML cell lines (Ba/F cells and their mutants highly resistant to high doses of imatinib mesylate) with or without imatinib mesylate- ST1571 (92). This signal transduction inhibitor - protein-tyrosine kinase inhibitor in patients with more advanced clinical phases often fail to respond that type of therapy. OSU-03012 sensitizes imatinib mesylate-resistant cells to imatinib-induced apoptosis irrespective to of Bcr-Abl mutation, which cause the chemotherapy resistance in advanced form of CML (92).
About twenty thousand of acute myeloid leukemia (AML) cases are diagnosed annually, which constitutes about 35% of all per annum leukemia cases in USA (59, 93-96). In a standard chemotherapy treatment of AML, anthracycline and cytarabine are commonly used. However, it has been reported that AML cells are frequently resistant to this treatment (97, 98). It was proved that the overexpression of COX-2 in tumour cells may cause inhibition of apoptosis but the exact mechanism of this phenomenon requires further studies (49). In an in vitro experiment with HL-60 cells, celecoxib prevented the autophagic flux by inhibiting lysosome function, as confirmed by upregulation of caspase-3 and PARP, as well as by apoptosis induction (99). Chen et al. demonstrated a pro-apoptotic effect of celecoxib and doxorubicin on the expression of survivin in human leukemia cell line HL-60 (49). It has also been shown that nimelisude down-regulates the IFN-γ induced expression of indoleamine 2,3-dioxygenase 1 (IDO 1) in HL-60 cells by gene transcription (100). This experiment also demonstrated that HL-60 cells stimulated by IFN-γ in the presence of nimelisude and then further incubated with allogenic CD4+CD25– T cell, transformed them into CD4+CD25+FoxP3+ regulatory T cells (100).
Acute lymphoblastic leukemias (ALLs) are as a result of an accumulation of mutations in tumour suppressor genes and oncogenes, and the genetic transformation affecting several chromosomes. ALLs represent 30% of all childhood leukemia but their exact etiology is still unknown (101-105). Wang et al. have investigated an association of 6 polymorphic variants of COX-2 in children with ALLs and have shown that the C allele of COX-2 G-765C may be responsible for ALLs in children (101). There are no data explaining the role of COX-2 in ALL cancerogenesis. In the veterinary literature there is no information about COX-2 expression and their influence on canine leukemic cells. Both studies suggest that COXIBs could enhance chemotherapeutic treatments in patients suffering from hematological malignancies. However, more clinical investigations are still required.
CYCLOOXYGENASE INHIBITORS IN CANCER TREATMENT AND PREVENTION
Non-steroidal anti-inflammatory drugs (NSAIDs) comprise a group of compounds that are used for relieving symptoms of inflammation. COXIBs administration prevents ulcering and gastrointestinal bleeding or perforation associated with prolonged use of non-specific NSAIDs but in many recent papers there has also been documented that they are implicated in carcinogenesis and have potential influence in metronomic chemotherapy of hematological malignancies. In one study, the median overall and progression-free survival times were measured during metronomic therapy with cyclophosphamide and celecoxib in patient with NHL (106). Plasma concentration of vascular endothelial growth factor, circulating endothelial cells (CECs) and circulating endothelial precursors cells (CEPs) were used as a markers of angiogenesis and they were increased in the blood of NHL patients (106-110). After cyclophosphamide and celecoxib therapy their number of CECs and CEPs in the blood significantly decreased while the median overall and progression-free survivals increased to 14.4 and 4.7 months, respectively, suggesting that this type of metronomic therapy may inhibit angiogenesis (106). COXIBs are now used in humans cancer treatment regimens of: colon, breast, pancreas, osteosarcoma, lung cancer and metronomic therapy and in individual cases of treatment regimens in dogs with: transitional cell carcinoma, inflammatory mammary carcinoma (18, 111-115).
In humans, dozens of epidemiological studies have examined a potential relation between frequent use of NSAIDs and the development of lymphoid malignancies, but the results report either an increased risk of NHL or decreased risk of these malignancies or no association. For example, the risk of lymphoid malignancies is reduced in patients who regularly used aspirin and other NSAIDs, while the frequent use of aspirin was found to be associated with increased risk of acute leukemia development (116). There are several possible explanations for the inconsistency in the results across the reported studies. NSAIDs are a diverse group of chemical agents and some patients were administrated two or more types of NSAIDs which could differently influence NHL development. There is only one study about the role of NSAIDs in the development and treatment of hematologic malignancies, while the influence of COXIBs in cancer is poorly described. One reason is that they are relatively recent drugs, especially in the treatment of hematologic malignancies. Studies also have shown lack of a full consistency in dose, frequency and duration of NSAIDs use, making direct comparisons of effects difficult (116, 117).
The use COXIBs in prevention or as adjunct therapy in patients with hematological malignancies seems to be reasonable, but at the same time lipoxygenase pathway or phospholipase A2 are not inhibited. Schain et al. in lymphoma cell line L1236 shows high expression of lipoxygenase 1 and confirm the ability to convert arachidonic acid to proinflammatory cytokines - eoxins. Another study shows, that only T cell-derived anaplastic large cell lymphoma shown expression of lipoxygenase-1 (118). COXIBs compared to traditional NSAIDs are considered relatively safe for gastrointestinal tract during long term administration, but may increase the risk of cardiovascular events. It has been demonstrated that COXIBs inhibit prostacyclin production in the vascular wall, which promote platelets aggregation and arteriosclerosis (12-14, 119). Moreover, inhibition of prostacyclin in the kidney lead to sodium and water retention, and could cause hypertension, and in long -term therapy might increase the risk of cardiovascular events (15, 119).
In this review the role of COX-2/PGE2 pathway in the regulation of tumour immune evasion has been characterized. The activity of COX-2 is suspected to promote angiogenesis, tissue invasion, resistance to apoptosis of cancer cells in many solid tumours and in hematological malignancies. This signalling pathway can suppress tumour immunity and act as an oncogene. Therefore, COXIBs are used with standard chemotherapy in some solid tumour treatment, which can help overcoming the tumour immune evasion. It is important to assess their potential influence in chemotherapy of hematological malignancies (9, 28, 40, 99, 100).
Abbreviations: ALLs, acute lymphoblastic leukemias; AML, acute myeloid leukemia; BL, Burkitt’s lymphoma; CECs, circulating endothelial cells; CEPs, circulating endothelial precursors cells; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; COX-2, cyclooxygenase-2; COXIBs, cyclooxygenase-2 inhibitors; CSCs, cancer stem cells; DLBCL, diffuse large B-cell lymphoma; EGR-1, early growth response factor 1; EP, prostaglandin receptors; ERKs, extracellular signal-regulated kinases; FL, follicular lymphoma; IDO 1, indoleamine 2,3-dioxygenase 1; IKK, IκB kinase; MDSCs, myeloid derived suppressor cells; NHLs, non-Hodgkin’s lymphomas; NSAIDs, nonsteroidal anti-inflammatory drugs; PGE2, prostaglandin E2; PGEs, prostaglandin synthases; PGG2, prostaglandin G2; PGH2, prostaglandin H2; PGs, prostaglandins; PI3K, phosphatidylinositide 3-kinase; PLTs, platelets; TR, therapeutic resistance; TRAIL, TNF-related apoptosis-inducing ligand.
Acknowledgements: The financial assistance of the Department of Pathology and Veterinary Diagnostic, Faculty of Veterinary Medicine, Warsaw University of Life Science (WULS SGGW), NCN (National Science Centre, Poland) MINIATURA grant number 2017/01/X/NZ5/01481 (to M.Z) and NCN (National Science Centre, Poland) grant number 2015/17/N/NZ5/00663 (to A.R.).
Conflict of interests: None declared.
REFERENCES
- Harris SG, Padilla J, Koumas L, Denise R Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol 2002; 23: 144-150.
- Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol 2012; 188: 21-28.
- Greenhough A, Smartt HJ, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009; 30: 377-386.
- Wu QB, Sun GP. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J Gastroenterol 2015; 21: 6206-6214.
- Carvalho MI, Pires I, Prada J, Ferreira AF, Queiroga FL. Positive interplay between CD3+ T-lymphocytes and concurrent COX-2/EGFR expression in canine malignant mammary tumors. Anticancer Res 2015; 35: 2915-2920.
- Chen EP, Markosyan N, Connolly E, et al. Myeloid cell COX-2 deletion reduces mammary tumor growth through enhanced cytotoxic T-lymphocyte function. Carcinogenesis 2014; 35: 1788-1797.
- Li Q, Liu L, Zhang Q, Liu S, Ge D, You Z. Interleukin-17 indirectly promotes M2 macrophage differentiation through stimulation of COX-2/PGE2 pathway in the cancer cells. Cancer Res Treat 2014; 46: 297-306.
- Gan L, Qiu Z, Huang J, et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int J Biol Sci 2016; 12: 1533-1543.
- Holt DH, Ma X, Kundu N, Fulton AM. Prostaglandin E2 suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol Immunother 2011; 60: 1577-1586.
- Muller C, Tufa DM, Chatterjee D, Muhlradt PF, Schmidt RE, Jacobs R. The TLR-2/TLR-6 agonist macrophage-activating lipopeptide-2 augments human NK cell cytotoxicity when PGE2 production by monocytes is inhibited by a COX-2 blocker. Cancer Immunol Immunother 2015; 64: 1175-1184.
- Liu B, Qu L, Yan S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int 2015; 15: 106. doi: 10.1186/s12935-015-0260-7
- Garcia-Recio M, Martinez-Serra J, Bento L, et al. Lenalidomide, celecoxib, and azacitidine therapy for blastic plasmocytoid dendritic cell neoplasm: a case report. Onco Targets Ther 2016; 9: 5507-5511.
- Bernard MP, Bancos S, Sime PJ, Phipps RP. Targeting cyclooxygenase-2 in hematological malignancies: rationale and promise. Curr Pharm Des 2008; 14: 2051-2060.
- Yaqub S, Henjum K, Mahic M, Jahnsen FL, Aandahl EM, Bjornbeth BA. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother 2008; 57: 813-821.
- Maturu P, Jones D, Ruteshouser EC, et al. Role of cyclooxygenase-2 pathway in creating an immunosuppressive microenvironment and in initiation and progression of Wilms’ tumor. Neoplasia 2017; 19: 237-249.
- Sharma S, Yang SC, Zhu L, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 2005; 65: 5211-5220.
- Yukawa T, Shimizu K, Maeda A, et al. Cyclooxygenase-2 genetic variants influence intratumoral infiltration of Foxp3-positive regulatory T cells in non-small cell lung cancer. Oncol Rep 2015; 33: 74-80.
- Dore M. Cyclooxygenase-2 expression in animal cancers. Vet Pathol 2011; 48: 254-265.
- Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 2013; 64: 409-421.
- Millanta F, Asproni A, Canale S, Citi S, Poli A. COX-2, mPGES-1 and EP2 receptor immunohistochemical expression in canine and feline malignant mammary tumours. Vet Comp Oncol 2016; 14: 270-280.
- Takeuchi K. Gastric cytoprotection by prostaglandin E2 and prostacyclin: relationship to EP1 and IP receptors. J Physiol Pharmacol 2014; 65: 3-14.
- Fujino H, Regan JW. Prostanoid receptors and phosphatidylinositol 3-kinase: a pathway to cancer? Trends Pharmacol Sci 2003; 24: 335-340.
- Cook PJ, Thomas R, Kingsley PJ, et al. COX-2-derived PGE2 induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro Oncol 2016; 18: 1379-1389.
- Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem 2001; 276: 18075-18081.
- Chang HH, Young SH, Sinnett-Smith J, et al. Prostaglandin E2 activates the mTORC1 pathway through an EP4/cAMP/PKA- and EP1/Ca2+-mediated mechanism in the human pancreatic carcinoma cell line PANC-1. Am J Physiol Cell Physiol 2015; 309: C639-C649.
- Sonoshita M, Takaku K, Sasaki N, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc (Delta716) knockout mice. Nat Med 2001; 7: 1048-1051.
- Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 2015; 149: 1884-1895.
- Xu L, Stevens J, Hilton MB, et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med 2014; 6: 242ra84. doi: 10.1126/scitranslmed.3008455
- Konturek PC, Konturek SJ, Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis. J Physiol Pharmacol 2009; 60: 3-21.
- Oksuz E, Atalar F, Tanirverdi G, Bilir A, Shahzadi A, Yazici Z. Therapeutic potential of cyclooxygenase-3 inhibitors in the management of glioblastoma. J Neurooncol 2016; 126: 271-278.
- Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA 2002; 99: 13926-13931.
- Munoz J, Navarro C, Noriega V, et al. Synergism between COX-3 inhibitors in two animal models of pain. Inflammopharmacology 2010; 18: 65-71.
- Tong D, Liu Q, Wang LA, et al. The roles of the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis Rev 2018; 37: 355-368.
- Veltman JD, Lambers ME, van Nimwegen M, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010; 10: 464. doi: 10.1186/1471-2407-10-464
- Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol 2012; 22: 282-288.
- Pingwara R, Witt-Jurkowska K, Ulewicz K, et al. Interferon lambda 2 promotes mammary tumor metastasis via angiogenesis extension and stimulation of cancer cell migration. J Physiol Pharmacol 2017; 68: 573-583.
- Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017; 8: 3649-3665.
- Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX-2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA 2017; 114: 1117-1122.
- Nakanishi Y, Kamijo R, Takizawa K, Hatori M, Nagumo M. Inhibitors of cyclooxygenase-2 (COX-2) suppressed the proliferation and differentiation of human leukaemia cell lines. Eur J Cancer 2001; 37: 1570-1578.
- Riva B, De Dominici M, Gnemmi I, et al. Celecoxib inhibits proliferation and survival of chronic myelogeous leukemia (CML) cells via AMPK-dependent regulation of b-catenin and mTORC1/2. Oncotarget 2016; 7: 81555-81570.
- Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol 2010; 2010: 215158. doi: 10.1155/2010/215158.
- Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am J Pathol 2001; 159: 387-397.
- Zeng W, Chang H, Ma M, Li Y. CCL20/CCR6 promotes the invasion and migration of thyroid cancer cells via NF-kappa B signaling-induced MMP-3 production. Exp Mol Pathol 2014; 97: 184-189.
- Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest 2001; 107: 135-142.
- Suthar SK, Sharma N, Lee HB, et al. Novel dual inhibitors of nuclear factor-kappa B (NF-κB) and cyclooxygenase- 2 (COX-2): synthesis, in vitro anticancer activity and stability studies of lantadene-non steroidal anti-inflammatory drug (NSAID) conjugates. Curr Top Med Chem 2014; 14: 991-1004.
- Arlt A, Gehrz A, Muerkoster S, et al. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003; 22: 3243-3251.
- Yip-Schneider MT, Nakshatri H, Sweeney CJ, Marshall MS, Wiebke EA, Schmidt CM. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer Ther 2005; 4: 587-594.
- Shin JS, Baek SR, Sohn SI, et al. Anti-inflammatory effect of pelubiprofen, 2-[4-(oxocyclohexylidenemethyl)-phenyl]propionic acid, mediated by dual suppression of COX activity and LPS-induced inflammatory gene expression via NF-κB inactivation. J Cell Biochem 2011; 112: 3594-3603.
- Chen C, Xu W, Wang CM. Combination of celecoxib and doxorubicin increases growth inhibition and apoptosis in acute myeloid leukemia cells. Leuk Lymphoma 2013; 54: 2517-2522.
- Martinez-Climent JA, Fontan L, Gascoyne RD, Siebert R, Prosper F. Lymphoma stem cells: enough evidence to support their existence? Haematologica 2010; 95: 293-302.
- Teng G, Dai Y, Chu Y, et al. Helicobacter pylori induces caudal-type homeobox protein 2 and cyclooxygenase 2 expression by modulating microRNAs in esophageal epithelial cells. Cancer Sci 2018; 109: 297-307.
- Yan M, Jurasz P. The role of platelets in the tumor microenvironment: from solid tumors to leukemia. Biochim Biophys Acta 2016; 1863: 392-400.
- Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med 2009; 206: 1913-1927.
- Yan M, Lesyk G, Radziwon-Balicka A, Jurasz P. Pharmacological regulation of platelet factors that influence tumor angiogenesis. Semin Oncol 2014; 41: 370-377.
- Baj-Krzyworzeka M, Majka M, Pratico D, et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp Hematol 2002; 30: 450-459.
- Dinkla S, van Cranenbroek B, van der Heijden WA, et al. Platelet microparticles inhibit IL-17 production by regulatory T cells through P-selectin. Blood 2016; 127: 1976-1986.
- Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 2015; 6: 364-372.
- Hu Q, Cho MS, Thiagarajan P, Aung FM, Sood AK, Afshar-Kharghan V. A small amount of cyclooxygenase 2 (COX2) is constitutively expressed in platelets. Platelets 2017; 28: 99-102.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7-30.
- Gallouet AS, Travert M, Bresson-Bepoldin L, et al. COX-2-independent effects of celecoxib sensitize lymphoma B cells to TRAIL-mediated apoptosis. Clin Cancer Res 2014; 20: 2663-2673.
- Landsburg DJ. Management of patients with MYC-altered lymphomas. Curr Hematol Malig Rep 2016; 11: 208-217.
- Mugnaini EN, Ghosh N. Lymphoma. Prim Care 2016; 43: 661-675.
- Phipps RP, Ryan E, Bernstein SH. Inhibition of cyclooxygenase-2: a new targeted therapy for B-cell lymphoma? Leuk Res 2004; 28: 109-111.
- Paul AG, Sharma-Walia N, Chandran B. Targeting KSHV/HHV-8 latency with COX-2 selective inhibitor nimesulide: a potential chemotherapeutic modality for primary effusion lymphoma. PLoS One 2011; 6: E24379. doi: 10.1371/journal.pone.0024379
- Jendrossek V, Handrick R, Belka C. Celecoxib activates a novel mitochondrial apoptosis signaling pathway. FASEB J 2003; 17: 1547-1549.
- Jendrossek V. Targeting apoptosis pathways by celecoxib in cancer. Cancer Lett 2013; 332: 313-324.
- Rudner J, Elsaesser SJ, Jendrossek V, Huber SM. Anti-apoptotic Bcl-2 fails to form efficient complexes with pro-apoptotic Bak to protect from celecoxib-induced apoptosis. Biochem Pharmacol 2011; 81: 32-42.
- Kobayashi M, Nakamura S, Shibata K, et al. Etodolac inhibits EBER expression and induces Bcl-2-regulated apoptosis in Burkitt’s lymphoma cells. Eur J Haematol 2005; 75: 212-220.
- Ben Abdelwahed Bagga R, Donnou S, Cosette J, Sautes-Fridman C, Aouni M, Fisson S. Mouse models of primary central nervous system lymphomas: tools for basing funding and therapeutic strategies. J Neurooncol 2015; 121: 9-18.
- Huang Y, Sheikh MS. TRAIL death receptors and cancer therapeutics. Toxicol Appl Pharmacol 2007; 224: 284-289.
- Gobec M, Prijatelj M, Delic J, Markovic T, Mlinaric-Rascan I. Chemo-sensitizing effects of EP4 receptor-induced inactivation of nuclear factor-kB. Eur J Pharmacol 2014; 742: 81-88.
- Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today 2017; 22: 57-71.
- O’Connor CM, Wilson-Robles H. Developing T cell cancer immunotherapy in the dog with lymphoma. ILAR J 2014; 55: 169-181.
- Rodrigues LC, Cogliati B, Guerra JL, Dagli ML, Lucas SR. An immunohistochemical study of cyclooxygenase-2 expression in canine multicentric lymphoma. An Vet (Muricia) 2011; 24: 43-47.
- Asproni P, Vignoli M, Cancedda S, Millanta F, Terragni R, Poli A. Immunohistochemical expression of cyclooxygenase-2 in normal, hyperplastic and neoplastic canine lymphoid tissues. J Comp Pathol 2014; 151: 35-41.
- Pang LY, Argyle SA, Kamida A, Morrison KO, Argyle DJ. The long-acting COX-2 inhibitor mavacoxib (Trocoxil™) has anti-proliferative and pro-apoptotic effects on canine cancer cell lines and cancer stem cells in vitro. BMC Vet Res 2014; 10: 184. doi:10.1186/s12917-014-0184-9.
- Pang LY, Gatenby EL, Kamida A, Whitelaw BA, Hupp TR, Argyle DJ. Global gene expression analysis of canine osteosarcoma stem cells reveals a novel role for COX-2 in tumour initiation. PLoS One 2014; 9: e83144. doi: 10.1371/journal.pone.0083144
- Rozovski U, Hazan-Halevy I, Keating MJ, Estrov Z. Personalized medicine in CLL: current status and future perspectives. Cancer Lett 2014; 352: 4-14.
- Lad D, Malhotra P, Varma N, et al. CLL: common leukemia; uncommon presentations. Indian J Hematol Blood Transfus 2016; 32: 268-725.
- Kaur V, Swami A. Ibrutinib in CLL: a focus on adverse events, resistance, and novel approaches beyond ibrutinib. Ann Hematol 2017; 96: 1175-1184.
- Pettijohn EM, Ma S. Targeted therapy in chronic lymphocytic leukemia (CLL). Curr Hematol Malig Rep 2017; 12: 20-28.
- Ryan EP, Pollock SJ, Kaur K, et al. Constitutive and activation-inducible cyclooxygenase-2 expression enhances survival of chronic lymphocytic leukemia B cells. Clin Immunol 2006; 120: 76-90.
- Rozovski U, Keating MJ, Estrov Z. Targeting inflammatory pathways in chronic lymphocytic leukemia. Crit Rev Oncol Hematol 2013; 88: 655-666.
- Secchiero P, Barbarotto E, Gonelli A, et al. Potential pathogenetic implications of cyclooxygenase-2 overexpression in B chronic lymphoid leukemia cells. Am J Pathol 2005; 167: 1599-1607.
- Johnson AJ, Smith LL, Zhu J, et al. A novel celecoxib derivative, OSU03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase- and Bcl-2-independent mechanism. Blood 2005; 105: 2504-2509.
- Cayssials E, Guilhot F. Chronic myeloid leukemia: immunobiology and novel immunotherapeutic approaches. BioDrugs 2017; 31: 143-149.
- Kandarpa M, Wu YM, Robinson D, Burke PW, Chinnaiyan AM, Talpaz M. Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofibrosis. Am J Hematol 2017; 92: 555-561.
- Litwinska Z, Machalinski B. miRNAs in chronic myeloid leukemia: small molecules, essential function. Leuk Lymphoma 2017; 58: 1297-1305.
- Dharmapuri G, Doneti R, Philip GH, Kalle AM. Celecoxib sensitizes imatinib-resistant K562 cells to imatinib by inhibiting MRP1-5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk Res 2015; 39: 696-701.
- Subhashini J, Mahipal SV, Reddanna P. Anti-proliferative and apoptotic effects of celecoxib on human chronic myeloid leukemia in vitro. Cancer Lett 2005; 224: 31-43.
- Peng HL, Zhang GS, Liu JH, Gong FJ, Li RJ. Dup-697, a specific COX-2 inhibitor, suppresses growth and induces apoptosis on K562 leukemia cells by cell-cycle arrest and caspase-8 activation. Ann Hematol 2008; 87: 121-129.
- Tseng PH, Lin HP, Zhu J, et al. Synergistic interactions between imatinib mesylate and the novel phosphoinositide-dependent kinase-1 inhibitor OSU-03012 in overcoming imatinib mesylate resistance. Blood 2005; 105: 4021-4027.
- Carneiro BA, Altman JK, Kaplan JB, et al. Targeted therapy of acute myeloid leukemia. Expert Rev Anticancer Ther 2015; 15: 399-413.
- Kantarjian H. Acute myeloid leukemia - major progress over four decades and glimpses into the future. Am J Hematol 2016; 91: 131-145.
- Taga T, Tomizawa D, Takahashi H, Adachi S. Acute myeloid leukemia in children: Current status and future directions. Pediatr Int 2016; 58: 71-80.
- Short NJ, Ravandi F. Acute myeloid leukemia: past, present, and prospects for the future. Clin Lymphoma Myeloma Leuk 2016; Suppl 16: S25-S29. doi: 10.1016/j.clml.2016.02.007
- Daver N, Cortes J, Kantarjian H, Ravandi F. Acute myeloid leukemia: advancing clinical trials and promising therapeutics. Expert Rev Hematol 2016; 9: 433-445.
- Hefti E, Blanco JG. Anthracycline-related cardiotoxicity in patients with acute myeloid leukemia and Down syndrome: a literature review. Cardiovasc Toxicol 2016; 16: 5-13.
- Lu Y, Liu XF, Liu TR, et al. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed Pharmacother 2016; 84: 1551-57.
- Iachininoto MG, Nuzzolo ER, Bonanno G, et al. Cyclooxygenase-2 (COX-2) inhibition constrains indoleamine 2,3-dioxygenase 1 (IDO1) activity in acute myeloid leukaemia cells. Molecules 2013; 18: 10132-10145.
- Wang CH, Wu KH, Yang YL, et al. Association study of cyclooxygenase 2 single nucleotide polymorphisms and childhood acute lymphoblastic leukemia in Taiwan. Anticancer Res 2010; 30: 3649-3653.
- Rubnitz JE, Hijiya N, Zhou Y, Hancock ML, Rivera GK, Pui CH. Lack of benefit of early detection of relapse after completion of therapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 2005; 44: 138-141.
- Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J 2017; 7: e577.
- Rytting ME, Jabbour EJ, O’Brien SM, Kantarjian HM. Acute lymphoblastic leukemia in adolescents and young adults. Cancer 2017; 123: 2398-2403.
- De Braekeleer M, Douet-Guilbert N, De Braekeleer E. Prognostic impact of p15 gene aberrations in acute leukemia. Leuk Lymphoma 2017; 58: 257-265.
- Buckstein R, Kerbel RS, Shaked Y, et al. High-dose celecoxib and metronomic „low-dose“ cyclophosphamide is an effective and safe therapy in patients with relapsed and refractory aggressive histology non-Hodgkin’s lymphoma. Clin Cancer Res 2006; 12: 5190-5198.
- Bertolini F, Paul S, Mancuso P, et al. Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res 2003; 63: 4342-4346.
- Blix ES, Husebekk A. Raiders of the lost mark - endothelial cells and their role in transplantation for hematologic malignancies. Leuk Lymphoma 2016; 57: 2752-2762.
- Marinaccio C, Nico B, Maiorano E, Specchia G, Ribatti D. Insights in Hodgkin lymphoma angiogenesis. Leuk Res 2014; 38: 857-861.
- Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A. The role of angiogenesis in human non-Hodgkin lymphomas. Neoplasia 2013; 15: 231-238.
- Spugnini EP, Porrello A, Citro G, Baldi A. COX-2 overexpression in canine tumors: potential therapeutic targets in oncology. Histol Histopathol 2005; 20: 1309-1312.
- Regulski M, Regulska K, Prukala W, Piotrowska H, Stanisz B, Murias M. COX-2 inhibitors: a novel strategy in the management of breast cancer. Drug Discov Today 2016; 21: 598-615.
- Guarnieri T. Non steroidal anti inflammatory drugs as gatekeepers of colon carcinoma highlight new scenarios beyond cyclooxygenases inhibition. Curr Cancer Drug Targets 2016; 16: 186-197.
- Hou LC, Huang F, Xu HB. Does celecoxib improve the efficacy of chemotherapy for advanced non-small cell lung cancer? Br J Clin Pharmacol 2016; 81: 23-32.
- Saito T, Tamura D, Asano R. Usefulness of selective COX-2 inhibitors as therapeutic agents against canine mammary tumors. Oncol Rep 2014; 31: 1637-1644.
- Robak P, Smolewski P, Robak T. The role of non-steroidal anti-inflammatory drugs in the risk of development and treatment of hematologic malignancies. Leuk Lymph 2008; 49: 1452-1462.
- Ye X, Casaclang N, Mahmud SM. Use of non-steroidal anti-inflammatory drugs and risk of non-Hodgkin lymphoma: a systematic review and meta-analysis. Hematol Oncol 2015; 33: 176-186.
- Schain F, Schain D, Mahshid Y, et al. Differential expression of cysteinyl leukotriene receptor 1 and 15-lipoxygenase-1 in non-Hodgkin lymphomas. Clin Lymphoma Myeloma 2008; 8: 340-7.
- Sooriakumaran P. COX-2 inhibitors and the heart: are all coxibs the same? Postgrad Med J 2006; 82: 242–245.
A c c e p t e d : October 30, 2018