Skeletal muscle possesses the unique capacity to consume energy at rates that are hundreds of times greater than resting values. This large range of energy demands presents the muscle cell with the challenge of maintaining an adequate energy state. One of the principle enzymes involved in preventing a precipitous decline in the cellular energy state at high energy demands is AMP deaminase (AMPD). The purpose of this review is to highlight the role that the enzyme AMPD plays in maintaining a viable energy state in skeletal muscle, particularly during conditions where energy demands temporarily outpace the capacity for energy supply.
Energy state
The principle function of skeletal muscle is to generate force and facilitate movement. The energy costs associated with maintaining muscle function and viability are supported by the hydrolysis of high energy phosphate bonds of ATP. The primary energy consumer in skeletal muscle during contractions is the myosin ATPase which accounts for approximately 70% of the total ATP consumed by skeletal muscle, while the remaining 25-30% of ATP hydrolysis supports the maintenance of the large calcium gradient between the cytoplasm and the sarcoplasmic reticulum (~1:10,000) via the sarcoplasmic reticulum ATPase (SERCA) (see review (1)). Thus, the vast majority of the energy from ATP hydrolysis during muscle contractions is devoted to the cellular work involved in both the actomyosin-ATPase and the maintenance of the calcium gradients necessary for the activation and deactivation of the myofilaments.
The capacity for the high energy phosphate bonds of ATP to provide energy for
the cellular work of maintaining ion gradients and the cross-bridge cycle is
defined by the Gibbs free energy of ATP hydrolysis. The Gibbs free energy of
a chemical reaction is a measure of the degree to which a reaction is from reaching
equilibrium (2). Simply put, the amount of free energy liberated from the ATP
hydrolysis reaction (ATP

ADP + P
i ) is a function of the relative concentrations
of ADP and inorganic phosphate (P
i) to ATP,
as defined below (equation 1).
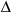
G
ATP=
G
oATP+RTln([ADP]*[P
i]/[ATP])
(equation 1)
The
G
oATP
is the free energy of ATP at defined standard conditions (-32 kJ/mol (3)), R
is the ideal gas constant, and T is the temperature in Kelvin. As the concentration
of ADP and P
i increase, and/or the concentration
of ATP decreases, the energy available from ATP hydrolysis declines. The actual
amount of energy available is particularly important since it may be limiting
for some ATPase reactions in the cell and, therefore, may effect muscle contractions.
The free energy available from ATP must be put in the context of both the content
and rate of ATP hydrolysis in skeletal muscle. The rate of ATP hydrolysis in
skeletal muscle can be as great as 10 µmol/g/s (3, 4) which would result in
the complete depletion of the existing content of ATP (~4-7 µmol/g) in less
than one second, in the absence of any ADP rephosphorylation. Fortunately, the
supply of ATP occurs rapidly and under most conditions is sufficient to maintain
adequate content of ATP for cellular viability. In fact, the rate of ATP hydrolysis
is matched by the rate of ATP supply without measurable changes in ATP over
a wide range of ATP demands (up to very intense short-term near-maximal exercise)
(5). The sources of ATP during sustained contractions are primarily oxidative
phosphorylation and to a smaller degree glycolysis. Additionally, phosphocreatine
(PCr)
via the creatine kinase reaction (reaction 1) also serves as a
limited source of high-energy phosphate, and as an intracellular buffer of ADP,
limiting the decline in ATP during transitions from low to high ATP demands.
PCr + ADP + ßH+ g Cr + ATP (reaction 1)
The importance of PCr and the creatine kinase reaction has been rigorously investigated with the use of models of PCr depletion as well as transgenic models of CK deficiency (6 - 12).
When ATP consumption is sufficiently out of balance with ATP supply, a decline
in ATP is observed, and this is precisely where the activity of the coupled
reactions of adenylate kinase (AK) and AMP deaminase (AMPD) are most important.
The primary effect of AK and AMPD on adenine nucleotide content is to limit
the accumulation of ADP when ATP turnover rates are high. This is particularly
clear during conditions when the rate of ATP is high enough to cause a net reduction
in ATP content. Consider for example, the fact that a 40% decline in ATP would
result in a 160 fold increase in ADP if all of the decline in ATP was converted
only to ADP (13). However, the range of metabolically available ADP concentrations
that have been reported in skeletal muscle is on the order of only 10 fold (~15
to 150 nmol/g muscle weight (13, 14)). It is important to note that similar
constraints on the concentration of P
i are not
observed, as the concentration of P
i largely
mirrors reductions observed in PCr and can reach concentrations near 20 µmol/g
muscle weight during high energy demands (15). Thus, limiting ADP accumulation
is critical to maintain an adequate ratio of ADP and P
i
to ATP and preserve sufficient energy available from ATP hydrolysis (equation
1). The manner in which ADP accumulation is limited by AK and AMPD is also closely
tied to the regulation of the concentration of AMP. The concentration of free
AMP in skeletal muscle is estimated to range from approximately 0.09 to 25 nmol/g
muscle weight (16). This is quantitatively insignificant (< 0.5%) relative to
the changes observed in ATP which can be ~3-4 µmol/g muscle weight. Thus, AK
and AMPD activity have a pivotal role in maintaining the free energy of ATP
hydrolysis.
During conditions when the rate of ATP hydrolysis outpaces the rate of ATP synthesis and a decline in ATP is observed, the AK reaction is critical in limiting an inordinate increase in the concentration of ADP. Adenylate kinase enzyme capacity in skeletal muscle is extremely high, and is considered to be near equilibrium under most conditions (17). The AK reaction consists of the phosphorylation of an ADP molecule by another ADP resulting in the formation of an ATP and an AMP (reaction 2). As alluded to above, the accumulation of AMP is also limited by the AMP deaminase reaction (reaction 3) which catalyzes the removal of an amino group from AMP forming IMP and ammonia. In contrast to AK, the reaction catalyzed by AMPD is irreversible at physiological condi-tions. Therefore, the recovery of adenine nucleotides lost during high energy demands must be recovered by the reamination of IMP. This occurs through the reactions of adenylosuccinate synthetase and adenylosuccinate lyase, which along with AMPD make up the reactions of the purine nucleotide cycle (see reviews (18 - 20).
2ADP

ATP + AMP (reaction 2)
AMP
+ H
2O
IMP + NH3 (reaction 3)
2ADP + H
2O
ATP + IMP +
NH
3 (reaction 4)
The high capacity of AMPD in skeletal muscle during the high energy demands
of intense muscle contractions keeps the AK reaction proceeding in the direction
of AMP formation and on balance, IMP formation. This has been illustrated in
rat skeletal muscle where the decline in ATP due to the high energy demands
of muscle contractions, is matched by an increase in IMP accumulation (
Fig.
1) (21 - 23). Furthermore, this also occurs in human muscle during intense
exercise bouts (24 - 26). Thus, when ATP hydrolysis outpaces supply, large accumulations
of ADP and AMP are preempted by the coupled reactions of AK and AMPD, facilitating
the accumulation of IMP and NH
3.
 |
Fig. 1. The decline in muscle
ATP content during intense contractions in fast-twitch muscle is matched
by a stoichiometric increase in IMP content. Adapted from Meyer, et
al., (22) with permission. |
Regulation of AMPD
While the enzymatic capacity of AMPD in skeletal muscle is extremely high, many
studies have demonstrated that AMPD is highly regulated. First, AMPD in skeletal
muscle is activated by a reduction in intracellular pH (27), and
in vitro
kinetics have demonstrated optimal activity at a pH of 6.5 (28). Second, AMPD
is allosterically regulated by ADP, AMP and P
i.
Increases in ADP and AMP concentrations have an activating effect, while increases
in P
i are inhibitory (29, 30). Third, AMPD binds
to myosin which causes an increase in AMPD activity (31 - 34). Furthermore,
an increase in AMPD binding to myosin occurs with muscle contractions and may
suggest that the localization of AMPD to sites of high ATP turnover is important
for effective enzyme activity (35, 36). Fourth, AMPD enzyme kinetics are enhanced
by phosphorylation (37), however the kinase(s) responsible for this activation
has yet to be fully characterized. Interestingly, the activation of AMPD in
skeletal muscle also requires the sustained ATP turnover of active contractions
even if the intracellular conditions would be expected to favor activation of
AMPD (low pH, high ADP and AMP) (38). Thus, the regulation of AMPD favors enzyme
activation during conditions of high energy demands and sustained ATP turnover.
The activation of AMPD and subsequent IMP accumulation is almost strictly a
characteristic of fast-twitch skeletal muscle. Work on rodent muscle has shown
that in the absence of severe ischemia and high rates of tetanic contractions,
significant accumulation of IMP
via AMPD does not occur in slow-twitch
high oxidative muscle (22, 23, 27, 39). This distinction between fast and slow-twitch
muscle is not solely due to differences in oxidative capacity. At high energy
demands ATP degradation and IMP accumulation is clearly apparent in high oxidative
fast-twitch muscle, which has higher oxidative capacity than slow twitch muscle
(23). Further, when energy demands are more moderate, fast-twitch red muscle
is more resistant to a loss of adenine nucleotides, consistent with the higher
capacity for ATP synthesis (21).
AMPD deficiency
Almost 30 years ago, a defect in AMPD activity was characterized in patients
with exercise related symptoms (40). Since then, AMPD deficiency has been characterized
as "the most common muscle enzyme defect in man" (41), since a defect in AMPD
activity is found in approximately 2% of muscle biopsies (42 - 44). The symptoms
associated with AMPD deficiency range from being asymptomatic to severe exertional
myalgia and other exercise related pain (43 - 46). In general, controlled studies
examining the consequences of AMPD deficiency have not reported a clear picture
relating AMPD deficiency and impaired muscle function. For example, in a study
by Norman
et al., healthy and AMPD deficient subjects were asked to perform
a high-intensity cycling test, which consists of a short explosive cycling bout
(47). Muscle power was measured during the test and biopsies were taken immediately
following the test in order to measure relevant metabolites. In this study,
no differences in mechanical power output were observed in AMPD deficient and
normal subjects, even though the IMP accumulation was only significant in the
normal controls (47). Another study by De Ruiter
et al. (48) examined
muscle function during repeated bouts of exercise in subjects with or without
AMPD deficiency and found mixed results. Five of the 8 subjects with AMPD deficiency
presented prolonged muscle relaxation, and were unable to complete the exercise
protocol while the remaining 3 AMPD deficient subjects exhibited no functional
difference from control subjects (48). These two studies illustrate that although
functional complications have been associated with AMPD deficiency, an absolute
functional impairment is not evident.
Potential consequences of a decline in the energy state
Functional consequences associated with AMPD deficiency have been attributed to the effect that elevated cellular ADP concentrations have on the cellular energy state. As alluded to above, intracellular ion gradients can be sensitive to changes in a reduction in energy available from ATP. For the purposes of this review, we will discuss the energy requirements of maintaining the calcium gradient in skeletal muscle as this has been found to be sensitive to a decline in energy from ATP, over physiological conditions.
The gradient between cytosolic calcium and the calcium sequestered in the sarcoplasmic
reticulum is ~1:10,000 at rest and as a result, the maintenance of this gradient
accounts for a large portion of skeletal muscle ATP turnover during contractions
(1, 49). The energy required to maintain the resting concentration of cytosolic
calcium is worth exploring for two reasons: first, if cytosolic calcium concentration
is not rapidly restored following a contraction, force generation will continue
resulting in muscle contracture; second, the maintenance of the resting cytosolic
concentration of calcium by the sarcoplasmic reticulum ATPase is thought to
be particularly sensitive to a fall in the energy available from ATP hydrolysis.
Work by Dawson, Gadian and Wilkie more than 25 years ago (50) examined the relationship
between the slowing of muscle relaxation and the calculated free energy of ATP
hydrolysis in frog gastrocnemius. In that study, they found a clear correlation
between the calculated decline in energy from ATP hydrolysis and the decline
in the rate constant of muscle relaxation (
Fig. 2) (50). Further, from
this correlation, the minimum energy from ATP required to obtain relaxation
could be extrapolated. The value determined as the minimum energy required to
obtain relaxation fit with other estimates of the cost of maintaining the resting
intracellular calcium gradient between the cytosol and the SR (-48 kJ/mol at
37°C (51)). Recent work has also shown that ADP has a more direct effect on
SERCA function in skeletal muscle. A study by Macdonald and Stevenson has shown
that ADP at concentrations near 1 mM causes leakage of calcium from the SR through
SERCA in fast-twitch muscle fibers, effectively reducing the capacity for SERCA
to maintain an adequate intracellular gradient (52). In addition, other work
by Tian and colleagues found that the cardiac contractile reserve was sensitive
to changes in the free energy available from ATP (53 - 55). Further, the impairment
in contractile reserve was the result of an inability to control intracellular
calcium. Thus, they hypothesized this impairment is caused by reduced SERCA
activity due to reduced energy available from ATP hydrolysis (53 - 55). Thus,
the regulation of intracellular calcium concentration has been found to be intimately
correlated with impaired contractile function and a decline in the energy available
from ATP. These studies demonstrate the importance of limiting inordinate increases
in metabolites that would result in less energy from the hydrolysis of ATP,
most notably P
i and/or ADP.
 |
| Fig. 2.
Muscle relaxation rate constant as a function of the energy available
from ATP (GATP).
Adapted from Dawson et al., (50) with permission. The original
data obtained at 4°C (see insert) have been recalculated to values expected
at 37°C (main panel). |
The consequences of not limiting large ADP accumulation during contractions
are not all necessarily a result of a decline in the cellular energy state.
ADP accumulation may impact on a fundamental process of skeletal muscle, the
actin and myosin cross-bridge cycle. Studies examining the effect of elevated
ADP concentrations on cross-bridge cycling have found that, if sufficiently
high, ADP can slow cross-bridge cycling (56, 57). The functional result of slowed
cross-bridge kinetics include, increased peak tension due to a higher number
of bound cross-bridges, slower rate of force development, and slowed relaxation
(52, 56, 58, 59). However, the range of ADP concentrations normally found
in
vivo is sufficiently small that an ADP dependent effect on cross-bridge
kinetics is not expected (14).
Transgenic AK deficiency as a model to examine AMPD
Recently, new evidence illustrating the relationship between muscle capacity
for AMP deamination and the protection of the cellular energy state has been
reported. Two studies by Hancock
et al., examined both the consequence
of adenylate kinase deficiency on ADP accumulation and muscle function during
high energy demands (60, 61). The model employed in these studies was the transgenic
knockout of the Adenylate kinase 1 isoform (62), which is highly expressed in
skeletal muscle. As a result of skeletal muscle AK deficiency, AMPD capacity
would be severely restricted, since AMP would not be produced by the transphosphorylation
of 2 ADPs. While AMPD1
-/- mice would be the most
direct means of establishing AMPD activity deficiency in muscle, development
of these animals has apparently been problematic. Therefore, the use of AK deficiency
in muscle is a valuable model to determine what energetic and functional consequences
would result in the context of AMPD deficiency. The AMP deamination capacity
of AK1 deficient muscle was challenged by eliciting tetanic contractions at
increasingly demanding contraction conditions (30, 60, 90 and 120 tetani/min)
in AK1 deficient and wild type mice. This was done to have a range of energy
demands in which to examine the role of AMPD in preserving muscle energy state.
As a result of the limited AK activity, AMP deamination during the high energy
demands of tetanic contractions was clearly limited as evidenced by markedly
reduced IMP accumulation. Diminished AMPD capacity resulted in an increased
accumulation of chemically measured ADP of approximately 0.90 µmol/g muscle
weight above the resting ADP concentration. This would represent an increase
in free ADP to ~1.5 mM, which is approximately 10 fold greater than what has
been estimated to occur in muscle with normal AK activity (
Fig. 3). Furthermore,
this increase in ADP was verified as existing in the 'free,' non-bound form
within the cell with
31P-NMR. This was the first
report of a direct measurement of free ADP in intact skeletal muscle (60). As
a result of this inordinate increase in ADP, the calculated energy from ATP
hydrolysis was severely impaired (-46 and -53 kJ/mol) in muscles from AK deficient
and WT muscles respectively (60, 61). Thus, these studies provide direct evidence
that limited AMPD capacity can result in ADP accumulation during extreme energy
demands of tetanic contractions.
 |
Fig. 3. The initial rate
of ADP accumulation in adenylate-1 knockout (AK-/-)
and wild type mice. Adapted from Hancock et al., (61) with permission. |
In addition to examining the metabolic impairment in AK deficient muscle, the
muscle function was also assessed. As pointed out above, restoring resting cytoplasmic
calcium concentrations following contraction is thought to be one of the most
sensitive processes to reductions in energy availability. Furthermore, if the
capacity of SERCA is sufficiently impaired, prolonged or absent muscle relaxation
would be expected to occur. In AK deficient muscles that had ADP accumulation
on the order of 1.5 mM a clear slowing of muscle relaxation was observed (
Fig.
4). Additionally the impaired relaxation kinetics were only evident at the
highest contraction frequencies examined (90 and 120 tetani/min). While relaxation
kinetics were clearly delayed, near complete relaxation was evident and overall
contractile function (force developed, rate of force development, and tension
time integral) was remarkably resistant to the expected consequences of a reduced
energy state and the high concentration of ADP.
 |
| Fig. 4.
Loss of muscle force during intense contraction conditions (120 tetani/min;
left panel) and a comparison of the contraction force profile between
the first and 40th contraction in the sequence for adenylate-1 knockout
(AK-/-) and wild type mice. The 40th contraction
was selected since muscle [ADP] was elevated to ~1.5 mM during this time.
Adapted from Hancock et al., (61) with permission. |
The cost of maintaining the calcium gradient between the cytosolic and sarcoplasmic
reticulum is defined by the relationship -2RT ln ([Ca
2+]sr/[Ca
2+]cyt).
Given the cytosolic calcium concentration (50-100 nM) is roughly one ten thousandth
of the calcium concentration in the SR (~1 mM), then the minimum energy required
to maintain this gradient is -51 to -48 kJ/mol. The increase in ADP in AK deficient
muscle, and the resulting challenge to the energy available from ATP (-46 kJ/mol)
would be expected to severely impair the capacity for calcium sequestration
(49, 50, 53). While relaxation was delayed, muscle contracture due to the inability
of SERCA to sequester calcium did not occur. One possible explanation for this
may be that the energy available was sufficient to achieve cytosolic calcium
concentration that was markedly higher than resting concentrations but low enough
where significant force production did not occur. For example, a calcium concentration
of 250 nM would not likely cause much force production (63) and the energy required
to achieve this cytosolic calcium concentration (assuming an [Ca
2+]sr
of 1 mM) would be -43 kJ/mol. Thus, sufficient energy exists to restore calcium
to levels near the threshold concentration for force generation even with the
large ADP accumulation that occurs in the absence of normal AK and AMPD activity.
Another possible reason for the surprisingly robust muscle function in the context
of such a large reduction in energy concerns the coupling efficiency of Ca
2+/ATP
by SERCA. The normal coupling of 2 Ca
2+/ATP
via
SERCA may be reduced when the ADP concentration is markedly elevated. As mentioned
above, a high concentration of ADP has been reported to cause calcium leak through
SERCA (52). This would effectively reduce the free energy required to sequester
each calcium ion, but increase the amount of ATP turnover by SERCA.
In conclusion, the high rates of ATP turnover possible in skeletal muscle can
temporarily exceed the capacity for ATP synthesis causing a net reduction in
ATP content. An inordinate accumulation of ADP is prevented when ATP depletion
occurs
via the AK and AMPD reactions, resulting in IMP accumulation that
matches losses in ATP. By limiting ADP accumulation, AMPD and AK function to
protect the cell from an excessive decline in the cellular energy state. If
AMP deamination is prevented during intense contractions and leads to sufficient
accumulation of ADP, there is expected to be a profound impact on muscle function.
However, even in the context of ADP concentrations near 1.5 mM, representing
a severe challenge to the energy state greater than previously observed, muscle
function was remarkably well maintained. Thus, the protection of
G
ATP
may not be an essential role of AMPD in fast-twitch skeletal muscle. This potentially
places a greater emphasis on the role of AMPD contributing to the retention
of adenine nucleotide pool and amino acid deamination within skeletal muscle.
Acknowledgements:
Cited work by the authors has been supported by NIH grants AR21617, AR43903.
National Biomedical Research Institute grant MA00210, and Michigan State University
grant IRPG 41006. C.R. Hancock is currently in the Department of Medicine, Washington
University School of Medicine, St. Louis, MO, supported by NIH grant T32 AG000078.
J.J. Brault is currently in the Department of Cell Biology, Harvard Medical
School, Boston, MA, supported by a National Space Biomedical Research Institute
grant.
REFERENCES
- Rall JA. Energetic aspects of skeletal muscle contraction: implications of fiber types. Exerc Sport Sci Rev 1985; 13: 33-74.
- Zubay GL. Biochemistry (4th ed). 1998; Dubuque, IA: Wm. C. Brown Pub.
- Meyer RA, Foley JM. Cellular processes integrating the metabolic response to exercise. In Handbook of Physiology: Section 12 Exercise: Regulation and Integration of Multiple Systems 1996; L.B. Rowell and J.T. Shepherd (eds), Am Physiol Soc pp. 841-869.
- Kushmerick MJ, Meyer RA, Brown TR. Regulation of oxygen consumption in fast- and slow-twitch muscle. Am J Physiol 1992; 263: C598-C606.
- Karlsson J, Saltin B. Lactate, ATP, and CP in working muscles during exhaustive exercise in man. J Appl Physiol 1970; 29: 596-602.
- Gorselink M, Drost MR, Coumans WA, van Kranenburg GP, Hesselink RP, van der Vusse GJ. Impaired muscular contractile performance and adenine nucleotide handling in creatine kinase-deficient mice. Am J Physiol 2001; 281: E619-E625.
- Gorselink M, Drost MR, van der Vusse GL. Murine muscle deficient in creatine kinase tolerate repeated series of high-intensity contractions. Pflugers Arch 2001; 443: 274-279.
- Roman BB, Foley JM, Meyer RA, Koretsky AP. Contractile and metabolic effects of increased creatine kinase activity in mouse skeletal muscle. Am J Physiol 1996; 270: C1236-C1245.
- Roman BB, Meyer RA, Wiseman RW. Phosphocreatine kinetics at the onset of contractions in skeletal muscle of MM creatine kinase knockout mice. Am J Physiol 2002; 283: C1776-C1783.
- Roman BB, Wieringa B, Koretsky AP. Functional equivalence of creatine kinase isoforms in mouse skeletal muscle. J Biol Chem 1997; 272: 17790-17794.
- Sweeney HL. The importance of the creatine kinase reaction: the concept of metabolic capacitance. Med Sci Sports Exerc 1994; 26: 30-36.
- van Deursen J, Heerschap A, Oerlemans F, et al. Skeletal muscles of mice deficient in muscle creatine kinase lack burst activity. Cell 1993; 74: 621-631.
- Kushmerick MJ, Moerland TS, Wiseman RW. Mammalian skeletal muscle fibers
distinguished by contents of phosphocreatine, ATP, and Pi.
Proc Natl Acad Sci USA 1992; 89: 7521-7525.
- Chase PB, Kushmerick MJ. Effect of physiological ADP concentrations on contraction of single skinned fibers from rabbit fast and slow muscles. Am J Physiol 1995; 268: C480-C490.
- Meyer RA, Brown TR, Krilowicz BL, Kushmerick MJ. Phosphagen and intracellular pH changes during contraction of creatine- depleted rat muscle. Am J Physiol 1986; 250: C264-C274.
- Tullson PC, Terjung RL. Adenine nucleotide degradation in striated muscle. Int J Sports Med 1990; 11: 47-55.
- Lawson JW, Veech RL. Effects of pH and free Mg2+
on the Keq of the creatine kinase reaction and other phosphate hydrolyses
and phosphate transfer reactions. J Biol Chem 1979; 254: 6528-3657.
- Lowenstein JM. Ammonia production in muscle and other tissues: the purine nucleotide cycle. Physiol Rev 1972; 52: 382-414.
- Lowenstein JM. The purine nucleotide cycle revisited. Int J Sports Med 1990; 11: 37-46.
- Terjung RL, Dudley GA, Meyer RA, Hood DA, Gorski J. Purine Nucleotide Cycle Function in Contracting Muscle. Biochemistry of Exercise 6th International Symposium 1986; B. Saltin, E. Richter and H. Galbo: Human Kinetics, pp. 131-147.
- Dudley GA, Terjung RL. Influence of aerobic metabolism on IMP accumulation in fast-twitch muscle. Am J Physiol 1985; 248: C37-C42.
- Meyer RA, Dudley GA, Terjung RL. Ammonia and IMP in different skeletal muscle fibers after exercise in rats. J Appl Physiol 1980; 49: 1037-1041.
- Meyer RA, Terjung RL. Differences in ammonia and adenylate metabolism in contracting fast and slow muscle. Am J Physiol 1979; 237: C111-C118.
- Sahlin K, Broberg S. Adenine nucleotide depletion in human muscle during exercise: causality and significance of AMP deamination. Int J Sports Med 1990; 11: 62-67.
- Sahlin K, Palmskog G, Hultman E. Adenine nucleotide and IMP contents of the quadriceps muscle in man after exercise. Pflugers Arch 1978; 374: 193-198.
- Zhao S, Snow RJ, Stathis CG, Febbraio MA, Carey MF. Muscle adenine nucleotide metabolism during and in recovery from maximal exercise in humans. J Appl Physiol 2000; 88: 1513-1519.
- Dudley GA, Terjung RL. Influence of acidosis on AMP deaminase activity in contracting fast- twitch muscle. Am J Physiol 1985; 248: C43-C50.
- Martini D, Ranieri-Raggi M, Sabbatini AR, Raggi A. Regulation of skeletal muscle AMP deaminase: lysine residues are critical for the pH-dependent positive homotropic cooperativity behaviour of the rabbit enzyme. Biochim Biophys Acta 2001; 1544: 123-132.
- Wheeler TJ, Lowenstein JM. Adenylate deaminase from rat muscle. Regulation by purine nucleotides and orthophosphate in the presence of 150 mM KCl. J Biol Chem 1979; 254: 8994-8999.
- Wheeler TJ, Lowenstein JM. Effects of pyrophosphate, triphosphate, and potassium chloride on adenylate deaminase from rat muscle. Biochemistry 1980; 19: 4564-2567.
- Shiraki H, Ogawa H, Matsuda Y, Nakagawa H. Interaction of rat muscle AMP deaminase with myosin. II. Modification of the kinetic and regulatory properties of rat muscle AMP deaminase by myosin. Biochim Biophys Acta 1979; 566: 345-352.
- Shiraki H, Ogawa H, Matsuda Y, Nakagawa H. Interaction of rat muscle AMP deaminase with myosin. I. Biochemical study of the interaction of AMP deaminase and myosin in rat muscle. Biochim Biophys Acta 1979; 566: 335-344.
- Rundell KW, Tullson PC, Terjung RL. Altered kinetics of AMP deaminase by myosin binding. Am J Physiol 1992; 263: C294-C299.
- Mahnke-Zizelman DK, Sabina RL. Localization of N-terminal sequences in human AMP deaminase isoforms that influence contractile protein binding. Biochem Biophys Res Commun 2001; 285: 489-495.
- Rundell KW, Tullson PC, Terjung RL. AMP deaminase binding in contracting rat skeletal muscle. Am J Physiol 1992; 263: C287-C293.
- Rundell KW, Tullson PC, Terjung RL. AMP deaminase binding in rat skeletal muscle after high-intensity running. J Appl Physiol 1993; 74: 2004-2006.
- Rush JWE, Tullson PC, Terjung RL. Molecular and kinetic alterations of muscle AMP deaminase during chronic creatine depletion. Am J Physiol 1998; 274: C465-C471.
- Sahlin K, Gorski J, Edstrom L. Influence of ATP turnover and metabolite changes on IMP formation and glycolysis in rat skeletal muscle. Am J Physiol 1990; 259: C409-C412.
- Tullson PC, Whitlock DM, Terjung RL. Adenine nucleotide degradation in slow-twitch red muscle. Am J Physiol 1990; 258: 258-265.
- Fishbein WN, Armbrustmacher VW, Griffin JL. Myoadenylate deaminase deficiency: a new disease of muscle. Science 1978; 200: 545-548.
- Gross M. Clinical heterogeneity and molecular mechanisms in inborn muscle AMP deaminase deficiency. J Inherit Metab Dis 1997; 20: 186-192.
- Fishbein WN. Myoadenylate deaminase deficiency: inherited and acquired forms. Biochem Med 1985; 33: 158-169.
- Mercelis R, Martin JJ, de Barsy T, Van den Berghe G. Myoadenylate deaminase deficiency: absence of correlation with exercise intolerance in 452 muscle biopsies. J Neurol 1987; 234: 385-389.
- Sabina RL. Myoadenylate deaminase deficiency. A common inherited defect with heterogeneous clinical presentation. Neurol Clin 2000; 18: 185-194.
- Kelemen J, Rice DR, Bradley WG, Munsat TL, DiMauro S, Hogan EL. Familial myoadenylate deaminase deficiency and exertional myalgia. Neurology 1982; 32: 857-863.
- Zollner N, Reiter S, Gross M, et al. Myoadenylate deaminase deficiency: successful symptomatic therapy by high dose oral administration of ribose. Klin Wochenschr 1986; 64: 1281-1290.
- Norman B, Sabina RL, Jansson E. Regulation of skeletal muscle ATP catabolism by AMPD1 genotype during sprint exercise in asymptomatic subjects. J Appl Physiol 2001; 91: 258-264.
- De Ruiter CJ, May AM, van Engelen BG, Wevers RA, Steenbergen-Spanjers GC, de Haan A. Muscle function during repetitive moderate-intensity muscle contractions in myoadenylate deaminase-deficient Dutch subjects. Clin Sci 2002; 102: 531-539.
- Hasselbach W, Oetliker H. Energetics and electrogenicity of the sarcoplasmic reticulum calcium pump. Annu Rev Physiol 1983; 45: 325-339.
- Dawson MJ, Gadian DG, Wilkie DR. Mechanical relaxation rate and metabolism studied in fatiguing muscle by phosphorus nuclear magnetic resonance. J Physiol 1980; 299: 465-484.
- Chen W, London R, Murphy E, Steenbergen C. Regulation of the Ca2+
gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F
nuclear magnetic resonance study. Circ Res 1998; 83: 898-907.
- Macdonald WA, Stephenson DG. Effects of ADP on sarcoplasmic reticulum function in mechanically skinned skeletal muscle fibres of the rat. J Physiol 2001; 532: 499-508.
- Tian R. Thermodynamic limitation for the sarcoplasmic reticulum Ca(2+)-ATPase
contributes to impaired contractile reserve in hearts. Ann N Y Acad Sci
1998; 853: 322-324.
- Tian R, Halow JM, Meyer M, et al. Thermodynamic limitation for Ca2+ handling contributes to decreased contractile reserve in rat hearts. Am J Physiol 1998; 275: C2064-C2071.
- Tian R, Ingwall JS. Energetic basis for reduced contractile reserve in isolated rat hearts. Am J Physiol 1996; 270: H1207-H1216.
- Cooke R, Pate E. The effects of ADP and phosphate on the contraction of muscle fibers. Biophys J 1985; 48: 789-798.
- Westerblad H, Dahlstedt AJ, Lannergren J. Mechanisms underlying reduced maximum shortening velocity during fatigue of intact, single fibres of mouse muscle. J Physiol 1998; 510: 269-277.
- Karatzaferi C, Myburgh KH, Chinn MK, Franks-Skiba K, Cooke R. Effect of an ADP analog on isometric force and ATPase activity of active muscle fibers. Am J Physiol 2003; 284: C816-C825.
- Tesi C, Piroddi N, Colomo F, Poggesi C. Relaxation kinetics following
sudden Ca(2+) reduction in single myofibrils
from skeletal muscle. Biophys J 2002; 83: 2142-2151.
- Hancock CR, Brault JJ, Wiseman RW, Terjung RL, Meyer RA. 31P-NMR
observation of free ADP during fatiguing, repetitive contractions of murine
skeletal muscle lacking AK1. Am J Physiol 2005; 288: C1298-C1304.
- Hancock CR, Janssen E, Terjung RL. Skeletal muscle contractile performance and ADP accumulation in adenylate kinase-deficient mice. Am J Physiol 2005; 288: C1287-C1297.
- Janssen E, Dzeja PP, Oerlemans F, et al. Adenylate kinase 1 gene deletion disrupts muscle energetic economy despite metabolic rearrangement. EMBO J 2000; 19: 6371-6381.
- Wetzel P, Gros G. Decay of Ca2+ and force
transients in fast- and slow-twitch skeletal muscles from the rat, mouse
and Etruscan shrew. J Exp Biol 1998; 201: 375-384.